📋 Key Information Summary
- Membranoproliferative glomerulonephritis (MPGN) is defined histologically by mesangial hypercellularity and capillary wall thickening, creating a 'double-contour' appearance.
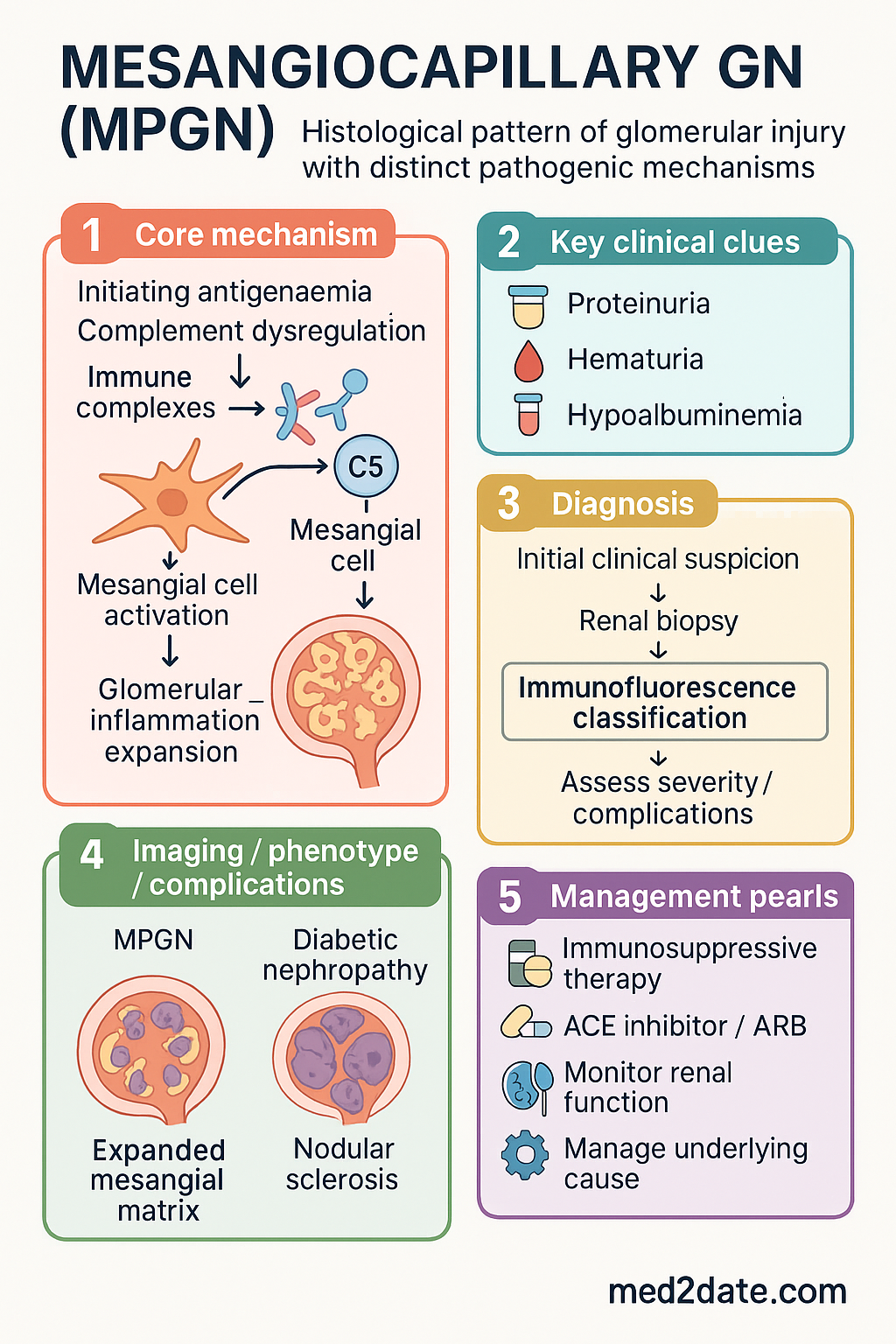
- The modern classification system divides MPGN into immune complex-mediated and complement-mediated subtypes, based on immunofluorescence findings.
- Immune complex MPGN is driven by chronic antigenaemia (e.g., infections like Hepatitis B/C, autoimmune disorders) leading to immune complex deposition.
- Complement-mediated MPGN is caused by dysregulation of the alternative complement pathway, often presenting as C3 glomerulopathy (C3GN or Dense Deposit Disease).
- Clinical presentation is heterogeneous: can range from asymptomatic haematuria/proteinuria to nephrotic or nephritic syndrome, or rapidly progressive glomerulonephritis (RPGN).
- Diagnosis requires renal biopsy with light, immunofluorescence, and electron microscopy to classify the subtype accurately.
- Key investigations include serum complement levels (C3, C4), infection screens (Hep B/C, HIV), autoimmune serology, and complement pathway functional assays.
- Treatment of immune complex MPGN targets the underlying cause (e.g., antiviral therapy for hepatitis).
- Treatment of complement-mediated MPGN/C3G is evolving; includes mycophenolate, corticosteroids, and complement inhibitors (e.g., eculizumab for specific cases).
- Supportive care with RAAS blockade and blood pressure control is critical for all forms.
- Prognosis is variable; significant risk of progression to end-stage kidney disease (ESKD), especially with nephrotic syndrome, heavy proteinuria, and renal impairment at presentation.
- ATSI populations may have a higher burden of chronic kidney disease; ensure culturally safe care and consider geographic barriers to specialist nephrology services.
Introduction & Australian Epidemiology
Membranoproliferative glomerulonephritis (MPGN), also termed mesangiocapillary glomerulonephritis, is a pattern of glomerular injury characterised histologically by mesangial hypercellularity and expansion, thickening of the capillary walls, and a duplication of the glomerular basement membrane (GBM), often giving a 'double-contour' appearance on silver stain. It is not a single disease but a pattern of injury resulting from several distinct pathogenic mechanisms.
The traditional classification based on electron microscopy appearance (Type I, II, III) has been largely superseded by a pathogenesis-based classification that divides MPGN into immune complex-mediated and complement-mediated disease. This reclassification has significant implications for investigation and targeted therapy.
In Australia, MPGN accounts for a minority of native kidney biopsies, but it remains an important cause of chronic kidney disease and nephrotic syndrome in both adults and children. Accurate epidemiological data are limited, but Australian biopsy series suggest that C3 glomerulopathy (a form of complement-mediated MPGN) is increasingly recognised, comprising a significant proportion of cases previously labelled as MPGN Type I. The incidence of infection-related MPGN has declined with hepatitis B vaccination and effective antiviral therapies.
Classification: Immune Complex vs Complement-Mediated
The modern classification of MPGN is based on the predominant driver of injury as identified by renal biopsy immunofluorescence (IF). This division is critical as it directs the diagnostic workup and therapeutic strategy.
| Feature | Immune Complex-Mediated MPGN | Complement-Mediated MPGN (C3 Glomerulopathy) |
|---|---|---|
| Primary Driver | Chronic antigenaemia leading to immune complex deposition. | Dysregulation of the alternative complement pathway. |
| Immunofluorescence | Dominant staining for immunoglobulins (IgG, IgM) and early complement components (C3, C1q, C4). | Dominant staining for C3 only (or C3 >2 intensity grades above any other reactant). |
| Common Causes | Chronic infections (Hepatitis B/C, HIV, endocarditis, shunt nephritis), autoimmune diseases (SLE, Sjögren's), monoclonal gammopathies. | Acquired (C3 nephritic factor, anti-Factor H antibodies) or genetic defects in complement regulators (Factor H, I, MCP). |
| Subtypes | Correlates with former MPGN Type I and III. | C3 Glomerulonephritis (C3GN): Electron-dense deposits in mesangium and subendothelium. Dense Deposit Disease (DDD): Highly electron-dense, sausage-shaped intramembranous deposits. |
| Serology Clue | Low C3 and C4 (if classical pathway activation). | Low C3 with normal C4 (isolated alternative pathway activation). |
Pathophysiology & C3 Glomerulopathy
Immune Complex MPGN
Persistent antigenaemia (e.g., from hepatitis B/C viral antigens, bacterial antigens in endocarditis) leads to sustained immune complex formation. These complexes deposit in the glomerulus, primarily in the subendothelial and mesangial spaces. This activates the classical complement pathway (hence low C3 and C4), inciting inflammation, mesangial cell proliferation, and leucocyte infiltration. The chronic process leads to GBM remodeling and the characteristic double contours.
C3 Glomerulopathy (C3G)
C3G is defined by the presence of dominant C3 staining on IF. The core defect is acquired or genetic dysregulation of the alternative complement pathway (AP).
- Acquired: Autoantibodies such as C3 nephritic factor (C3NeF) stabilise the AP C3 convertase, leading to uncontrolled C3 consumption and deposition. Anti-Factor H antibodies can also impair complement regulation.
- Genetic: Mutations in genes encoding complement regulatory proteins (Factor H, Factor I, MCP/CD46) or in amplification proteins (C3, Factor B) predispose to overactivation.
The resulting C3 fragments and membrane attack complex (MAC) deposit in the glomerulus, driving inflammation and injury. In Dense Deposit Disease (DDD), the deposited material is uniquely electron-dense, possibly due to the composition of C3 fragments.
Clinical Features & Natural History
Presentation is variable and does not reliably distinguish the underlying subtype.
Nephritic: Haematuria (dysmorphic RBCs, casts), hypertension, reduced GFR, mild-moderate proteinuria.
Natural History: The disease is typically chronic and progressive. Approximately 40-50% of patients with MPGN progress to ESKD within 10-15 years of diagnosis. Poor prognostic indicators include heavy proteinuria (>3.5 g/day), renal impairment (elevated creatinine) at presentation, and crescents on biopsy. DDD has a high recurrence rate post-transplant.
Investigations & Management
Diagnostic Investigations
Management Principles
Management is tailored to the underlying cause (immune complex vs complement) and clinical presentation.
1. Supportive Care (All Patients)
Aim for blood pressure <130/80 mmHg. Use loop diuretics for oedema. Manage cardiovascular risk factors.
2. Immunosuppressive Therapy
Indicated for nephrotic syndrome, RPGN, or progressive decline in GFR. No universal standard; therapy is often based on case series and expert opinion.
- Immune Complex MPGN: Treat underlying cause first (e.g., antivirals for HBV/HCV). Immunosuppression (e.g., mycophenolate mofetil ± corticosteroids) may be used for primary disease or if nephrotic syndrome persists after treating the cause.
- C3 Glomerulopathy: First-line often includes mycophenolate mofetil with corticosteroids. Consider eculizumab (PBS Authority Required) in severe cases, especially with crescents or post-transplant recurrence, particularly if genetic testing identifies a complement mutation or high-titre C3NeF.
3. Plasma Exchange
May be considered in specific scenarios: anti-Factor H antibody-mediated disease, severe crescentic GN, or in preparation for transplant in patients with known pathogenic antibodies.
Special Populations
High risk of pre-eclampsia and fetal loss. Requires joint obstetric-nephrology care.
MPGN in children is often idiopathic. Post-streptococcal GN must be excluded. DDD can present with partial lipodystrophy. Complement studies are essential. Dosing of immunosuppressants requires specialist paediatric nephrology guidance.
Dose-adjust medications. Renal impairment at presentation is a poor prognostic sign. Manage ESKD as per standard protocols. Plan for renal replacement therapy and transplant assessment early.
Exclude infections (HIV, HCV) as a cause. Monitor for opportunistic infections if on immunosuppression. Ensure vaccinations are up-to-date prior to therapy.
ATSI Health Considerations
📚 References
- 1. Kidney Disease: Improving Global Outcomes (KDIGO) Glomerular Diseases Work Group. KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int. 2021;100(4):S1-S276.
- 2. Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis – a new classification. Nat Rev Nephrol. 2012;8(10):611-622.
- 3. Pickering MC, D'Agati VD, Nester CM, et al. C3 glomerulopathy: consensus report. Kidney Int. 2013;84(6):1079-1089.
- 4. Australian Institute of Health and Welfare (AIHW). Chronic kidney disease: Australian facts. AIHW; 2023.
- 5. Therapeutic Goods Administration (TGA). Product Information: Eculizumab (Soliris®). 2022.
- 6. Rangan GK, Alexander SI, Campbell V, et al. KHA-CARI guideline recommendations for the diagnosis and management of glomerulonephritis. Nephrology. 2020;25(Suppl 1):1-103.
- 7. Cook HT, Pickering MC. Histopathology of MPGN and C3 glomerulopathies. Nat Rev Nephrol. 2015;11(1):14-22.
- 8. Smith RJH, Appel GB, Blom AM, et al. C3 glomerulopathy – understanding a rare complement-driven renal disease. Nat Rev Nephrol. 2019;15(3):129-143.
- 9. Rajagopalan R, Brailey MJ, Jones EA, et al. The burden of membranoproliferative glomerulonephritis: a systematic review. BMC Nephrol. 2021;22(1):199.
- 10. National Kidney Foundation (NKF). KDOQI Clinical Practice Guideline for Diabetes and CKD: 2022 Update. Am J Kidney Dis. 2022;80(4):S1-S127. [Note: For supportive care principles applicable to CKD from any cause, including MPGN].