📋 Key Information Summary
- Minimal change disease (MCD) is the most common cause of nephrotic syndrome in children (80–90%) and accounts for 10–15% of adult nephrotic syndrome cases in Australia.
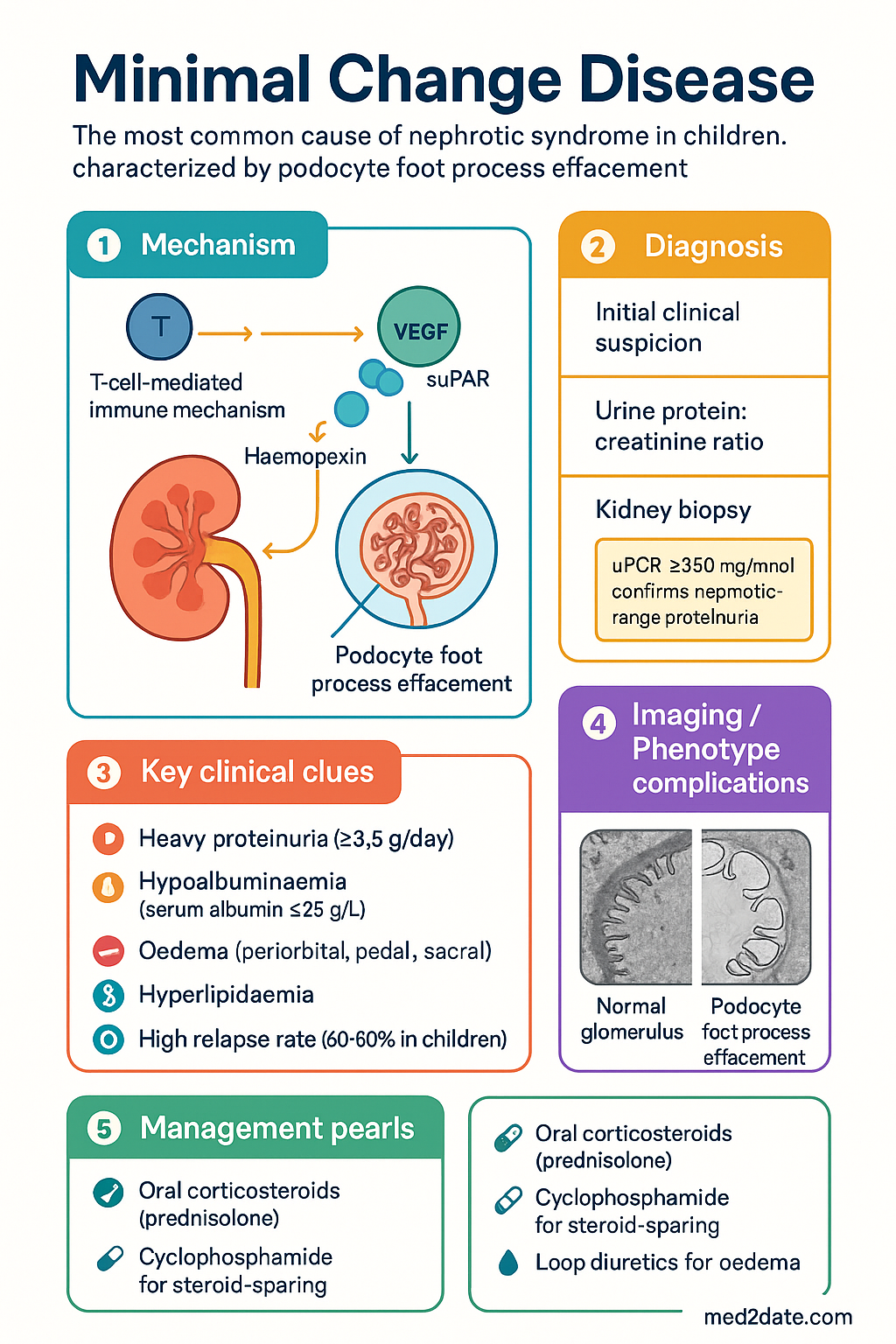
- MCD is characterised by diffuse podocyte foot process effacement on electron microscopy (EM) with normal light microscopy (LM) and negative immunofluorescence.
- Nephrotic syndrome presentation: heavy proteinuria (≥3.5 g/day or urine protein:creatinine ratio ≥350 mg/mmol), hypoalbuminaemia, oedema, and hyperlipidaemia.
- Over 90% of children and 70–80% of adults respond to oral corticosteroids — typically prednisolone — making MCD the most steroid-responsive glomerulopathy.
- Children usually respond within 4 weeks; adults may require up to 16 weeks of initial steroid therapy before classifying as steroid-resistant.
- Relapse rate is high (60–90% in children, 50–70% in adults); frequently relapsing (FRNS) and steroid-dependent (SDNS) patterns are common.
- Oral cyclophosphamide is the second-line steroid-sparing agent for FRNS/SDNS in both children and adults; calcineurin inhibitors (ciclosporin, tacrolimus) are alternatives.
- Supportive care with loop diuretics (furosemide ± spironolactone), sodium restriction, and compression stockings for oedema management.
- Thromboprophylaxis with low-molecular-weight heparin (LMWH) is recommended for adults with serum albumin <25 g/L due to increased thromboembolic risk.
- ACE inhibitors or ARBs should be initiated to reduce proteinuria and provide renoprotection once the acute nephrotic episode is controlled.
- Secondary MCD must be excluded — drug-induced (NSAIDs, lithium, interferon), malignancy (Hodgkin lymphoma), and infection-associated aetiologies.
- Kidney biopsy is essential in all adults and in children who are steroid-resistant or relapsing <1 year after first presentation.
- Infection is the leading cause of death in paediatric MCD — pneumococcal and varicella infections carry significant risk; immunisation status must be maintained.
Introduction & Australian Epidemiology
Minimal change disease (MCD), also termed minimal change nephropathy or nil disease, is the most common cause of nephrotic syndrome in childhood and a clinically significant cause in adulthood. It is characterised by the nephrotic syndrome — heavy proteinuria, hypoalbuminaemia, oedema, and hyperlipidaemia — with the distinctive histopathological finding of diffuse podocyte foot process effacement on electron microscopy, while light microscopy appears essentially normal and immunofluorescence is negative.
The hallmark of MCD is its excellent response to corticosteroid therapy, with complete remission rates exceeding 90% in children and 70–80% in adults. However, relapse is frequent, and many patients require multiple courses of immunosuppression over their lifetime. MCD is generally associated with a favourable long-term renal prognosis, progressing to end-stage kidney disease (ESKD) in fewer than 5% of cases, though the morbidity from relapsing nephrotic syndrome, treatment side effects, and complications is substantial.
| Epidemiological Feature | Detail |
|---|---|
| Childhood nephrotic syndrome cause | 80–90% of cases |
| Adult nephrotic syndrome cause | 10–15% of cases |
| Peak paediatric incidence | 2–6 years; male predominance (2:1) |
| Adult presentation age | All ages; mean 30–40 years |
| Steroid response rate (children) | >90% |
| Steroid response rate (adults) | 70–80% |
| Relapse rate | 60–90% children; 50–70% adults |
| Progression to ESKD | <5% |
Pathophysiology & Associations
Pathogenesis
The fundamental lesion in MCD is diffuse effacement (flattening) of the podocyte foot processes — the interdigitating extensions of glomerular visceral epithelial cells that form the slit diaphragm, a critical component of the glomerular filtration barrier. Loss of foot process architecture leads to increased permeability of the glomerular basement membrane (GBM) to albumin and other plasma proteins, producing selective proteinuria dominated by albumin.
The aetiology of podocyte injury in primary (idiopathic) MCD remains incompletely understood. Current evidence supports a T-cell-mediated immune mechanism producing one or more circulating permeability factors ("circulating factors") that directly injure podocytes. The rapid response to corticosteroids — often within days — and association with Hodgkin lymphoma and T-cell lymphomas are consistent with this immune-mediated pathogenesis. Proposed factors include haemopexin, vascular endothelial growth factor (VEGF), cardiotrophin-like cytokine factor 1 (CLCF-1), and soluble urokinase plasminogen activator receptor (suPAR), though none has been definitively established.
Additional pathogenic mechanisms under investigation include:
- Podocyte cytoskeletal dysregulation: Altered expression of synaptopodin, nephrin, and podocin leading to impaired slit diaphragm integrity.
- B-cell involvement: Recent evidence of podocyte autoantibodies (anti-nephrin) suggests a humoral component alongside T-cell dysregulation.
- Toll-like receptor activation: Innate immune triggers (viral infections) may initiate relapse via TLR signalling on podocytes and immune cells.
- Cytokine milieu: Interleukin-13 (IL-13) overproduction has been linked to podocyte injury and altered glomerular charge selectivity.
Secondary Causes & Associations
While most cases are idiopathic (primary MCD), secondary forms must be actively excluded, particularly in adults and atypical presentations.
| Category | Specific Associations |
|---|---|
| Drugs | NSAIDs (most common), lithium, interferon-α, bisphosphonates (pamidronate), penicillamine, mercury (skin-lightening creams), rifampicin |
| Malignancy | Hodgkin lymphoma (strongest association), non-Hodgkin lymphoma, thymoma, renal cell carcinoma |
| Infections | HIV, syphilis, hepatitis B (rare), Mycoplasma |
| Allergic / Atopic | Eczema, allergic rhinitis, food allergy (historical association, mechanism unclear) |
| Other | Graft-versus-host disease post-allogeneic stem cell transplant, vaccinations (rare temporal association) |
Clinical Features (Nephrotic Syndrome)
Defining Nephrotic Syndrome
MCD typically presents with the full or partial nephrotic syndrome, defined by the following cardinal features:
Clinical Presentation
Children typically present between ages 2–6 years with insidious onset of periorbital oedema, often worse in the morning and precipitated by intercurrent viral infections. Boys are affected twice as frequently as girls before puberty. The oedema may progress rapidly to generalised anasarca, with ascites, scrotal or vulval swelling, and pleural effusions.
Adults present at any age with gradual onset of dependent oedema, weight gain, and frothy urine. Presentation is less dramatic than in children, and proteinuria may be detected incidentally on routine urinalysis. Adults more commonly present with microscopic haematuria (20–30% vs. <20% in children) and are more likely to have hypertension (30–40%) and renal impairment (20–30%) at diagnosis.
Characteristic Features of MCD Nephrotic Syndrome
- Selective proteinuria: Predominantly albumin (high selectivity index) — more common in MCD than in other glomerulopathies.
- Absence or mild haematuria: Microscopic haematuria occurs in <20% of children and 20–30% of adults; gross haematuria is rare and should prompt consideration of IgA nephropathy or FSGS.
- Normal or near-normal GFR: eGFR is typically preserved; transient acute kidney injury may occur with severe hypovolaemia or diuretic overuse.
- Hypertension: Present in 10–20% of children, 30–40% of adults at presentation.
- Hyperlipidaemia: Elevated total cholesterol and LDL; triglycerides variably elevated. Secondary to hepatic lipoprotein synthesis in response to hypoalbuminaemia.
- Hypercoagulability: Particularly when serum albumin <20–25 g/L; renal vein thrombosis, deep vein thrombosis, pulmonary embolism, and cerebral venous sinus thrombosis (especially in children) are recognised complications.
Complications of Nephrotic Syndrome
| Complication | Mechanism / Notes |
|---|---|
| Infection | Urinary loss of immunoglobulins (especially IgG), complement factors; peritonitis (Spontaneous Bacterial Peritonitis — pneumococcus most common); cellulitis; sepsis. Leading cause of death in paediatric MCD. |
| Thromboembolism | Renal vein thrombosis (5–10% in adults), DVT, PE, cerebral venous thrombosis (children). Risk highest with albumin <20 g/L. |
| Hyperlipidaemia | Accelerated atherosclerosis risk with prolonged nephrotic syndrome; statin therapy (atorvastatin) considered if persistently elevated after 3 months of nephrotic state. |
| Acute kidney injury | Hypovolaemia, diuretic overuse, NSAIDs, ACE inhibitor initiation during severe oedema; usually reversible. |
| Protein malnutrition | Negative nitrogen balance despite adequate intake; muscle wasting in prolonged nephrotic state. |
| Vitamin D deficiency | Urinary loss of vitamin D binding protein; monitor 25-OH vitamin D levels and supplement as needed. |
| Growth retardation (children) | Chronic disease, steroid use, nutritional losses; monitor growth velocity. |
Investigations
Investigation of suspected MCD should confirm nephrotic syndrome, exclude secondary causes, assess complications, and confirm the diagnosis histologically where indicated.
Laboratory Investigations
Kidney Biopsy
Renal biopsy is the definitive diagnostic investigation and is required in all adults with new-onset nephrotic syndrome. In children, empirical steroid therapy is often commenced without biopsy for first episode, with biopsy reserved for steroid resistance, frequent relapse, or atypical features.
Histopathological Findings in MCD
| Modality | Findings |
|---|---|
| Light Microscopy (LM) | Normal or near-normal glomerular architecture. No proliferation, no thickening of GBM, no sclerosis. Occasional mild mesangial hypercellularity or increased mesangial matrix may be seen but is not prominent. Tubular epithelial cells may show reabsorption droplets (protein resorption droplets) in the cytoplasm. |
| Immunofluorescence (IF) | Negative for all immunoglobulins (IgG, IgA, IgM) and complement (C3, C1q). Weak IgM positivity in mesangium may be seen in a minority of cases and may predict steroid resistance. |
| Electron Microscopy (EM) | Diagnostic — diffuse podocyte foot process effacement. Complete flattening of foot processes (villous transformation) covering >80% of the GBM surface area. No electron-dense deposits. GBM thickness is normal. Mesangial areas may show mild increase in matrix. This is the key differentiating feature from early FSGS (where foot process effacement is also present but focal segmental sclerosis is visible on LM). |
Management
Management of MCD involves initial remission induction with corticosteroids, management of nephrotic syndrome complications, and long-term strategies to minimise relapse and steroid toxicity. The treatment approach varies between children and adults, and between first episode and relapsing disease.
Treatment Definitions
| Term | Definition |
|---|---|
| Complete remission | uPCR <50 mg/mmol (or urine protein <0.3 g/day) for 3 consecutive days |
| Partial remission | uPCR 50–200 mg/mmol with serum albumin improvement |
| Steroid resistance | Failure to achieve complete remission after 16 weeks (adults) or 4 weeks (children) of daily prednisolone |
| Relapse | uPCR >200 mg/mmol (or ≥3+ on dipstick) after having achieved remission |
| Frequently relapsing (FRNS) | ≥2 relapses within 6 months of initial response, or ≥4 relapses in any 12-month period |
| Steroid dependent (SDNS) | Relapse during steroid taper or within 2 weeks of stopping steroids |
First-Line Therapy: Corticosteroids
Management of Complications
Second-Line Therapy: Steroid-Sparing Agents for FRNS/SDNS
Treatment Algorithm
Monitoring
During Active Nephrotic Syndrome
During Steroid Therapy
| Parameter | Frequency |
|---|---|
| Blood glucose (fasting or HbA1c) | Every 3 months; more frequently if risk factors |
| Blood pressure | Every visit |
| Weight / BMI | Every visit |
| Ophthalmology review | Baseline, then annually if cumulative >6 months exposure |
| Bone density (DEXA) | If cumulative prednisolone >3 months; repeat every 1–2 years |
| 25-OH Vitamin D | Baseline and every 6 months; supplement if <50 nmol/L |
| Growth (children) | Height velocity every 3–6 months; plot on growth chart |
During Cyclophosphamide Therapy
Long-Term Follow-Up
- Nephrology review every 3–6 months during stable remission; more frequently during relapse or active taper.
- Annual urine protein assessment (uPCR), serum creatinine/eGFR, lipids, and blood pressure.
- Maintain lifelong awareness of relapse risk — patients should have urine dipstick testing available at home and know to seek medical review for frothy urine, oedema, or weight gain >1 kg/day.
- Annual influenza vaccination (funded under NIP); pneumococcal vaccination (PCV13 + PPSV23) and varicella vaccination (if non-immune) are essential.
- Avoid nephrotoxic agents: NSAIDs, aminoglycosides, iodinated contrast (unless essential with hydration protocol).
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Kidney Disease: Improving Global Outcomes (KDIGO). KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int. 2021;100(4S):S1–S276.
- 2. Cattran DC, Brenchley PE. Minimal change disease. In: Feehally J, Floege J, Tonelli M, Johnson RJ, eds. Comprehensive Clinical Nephrology. 6th ed. Philadelphia: Elsevier; 2019:285–298.
- 3. Vivarelli M, Massella L, Ruggiero B, Emma F. Minimal change disease. Clin J Am Soc Nephrol. 2017;12(2):332–345.
- 4. Australian Institute of Health and Welfare (AIHW). Chronic kidney disease: Australian facts