📋 Key Information Summary
- Neuromuscular weakness encompasses disorders of the anterior horn cell, peripheral nerve, neuromuscular junction, and muscle — each producing recognisable clinical patterns that guide diagnosis in primary care.
- Proximal weakness (difficulty rising from a chair, climbing stairs, overhead tasks) suggests myopathy or neuromuscular junction disorder; distal weakness (foot drop, grip weakness) points to peripheral neuropathy or motor neuron disease.
- Areflexia or hyporeflexia with acute or subacute weakness is a red flag for Guillain–Barré syndrome (GBS) and requires urgent hospital referral.
- Fatigability — weakness worsening with repeated use and improving with rest — is the hallmark of myasthenia gravis (MG) and should prompt acetylcholine receptor antibody testing.
- Respiratory red flags — dyspnoea at rest, orthopnoea, weak cough, difficulty swallowing, nasal speech — may indicate impending respiratory failure in GBS or myasthenic crisis; these patients require emergency department assessment and ICU-level monitoring.
- Guillain–Barré syndrome presents as rapidly progressive bilateral flaccid weakness with areflexia, often ascending from the legs; nerve conduction studies confirm demyelination; IV immunoglobulin (IVIg) or plasma exchange are first-line treatments.
- Myasthenic crisis is a life-threatening exacerbation causing respiratory failure; requires intubation, IVIg or plasma exchange, and ICU admission. Distinguish from cholinergic crisis clinically and with edrophonium testing or ice-pack test.
- Spinal cord compression — acute bilateral leg weakness, sensory level, urinary retention, and bowel dysfunction — is a neurological emergency requiring emergent MRI and decompression within 24 hours.
- Inflammatory myopathies (polymyositis, dermatomyositis, inclusion body myositis) present with subacute proximal weakness and elevated CK; anti-Jo-1, anti-Mi-2, and anti-SRP antibodies guide classification and prognosis.
- Hereditary myopathies (facioscapulohumeral dystrophy, limb-girdle dystrophy, myotonic dystrophy) typically present with insidious onset, family history, and characteristic patterns such as facial weakness or myotonia.
- Baseline investigations for suspected myopathy include serum CK, ESR/CRP, thyroid function, serum electrolytes (potassium, calcium, magnesium), autoimmune screen (ANA, ENA, myositis-specific antibodies), and HbA1c for statin-related myopathy.
- Aboriginal and Torres Strait Islander Australians may present later due to barriers to specialist access; culturally safe screening, awareness of delayed presentations, and supported referral pathways are essential.
- Muscle biopsy, electromyography (EMG), and nerve conduction studies (NCS) are available in major Australian centres; telehealth and fly-in specialist services support regional and remote assessment.
Introduction & Australian Epidemiology
Neuromuscular weakness refers to impairment of strength arising from disease at any point along the motor pathway: the anterior horn cells (motor neuron disease), peripheral nerves (neuropathies including GBS), the neuromuscular junction (myasthenia gravis, Lambert–Eaton syndrome), or the muscle itself (myopathies and muscular dystrophies). Accurate localisation is the critical first step in evaluation, and the pattern of weakness — proximal versus distal, symmetric versus asymmetric, with or without sensory involvement — provides the essential clue.
In Australia, neuromuscular disorders collectively affect tens of thousands of individuals. Myasthenia gravis has an estimated prevalence of approximately 150–250 per million population, with a bimodal age of onset peaking in women aged 20–30 and men aged 60–80. Guillain–Barré syndrome occurs in approximately 1–2 per 100,000 person-years, with a mean age of onset around 40 years. Inflammatory myopathies have an incidence of approximately 5–10 per million per year, while hereditary muscular dystrophies such as myotonic dystrophy type 1 affect approximately 1 in 8,000 Australians. Motor neuron disease (MND) has an incidence of approximately 2–3 per 100,000 per year, with a median survival of 2–3 years from diagnosis.
Aboriginal and Torres Strait Islander Australians may experience higher rates of diabetes-related neuropathy and delayed diagnosis of neuromuscular conditions due to geographic isolation and barriers to specialist access. National surveillance data from the Australian Institute of Health and Welfare (AIHW) and the Australian Neuromuscular Disease Registry highlight the need for improved access to diagnostic services in rural and remote areas.
Screening in Primary Care
The general practitioner is often the first clinician to evaluate a patient with neuromuscular weakness. A structured approach to history and examination allows rapid identification of the localisation (anterior horn cell, nerve, junction, muscle), urgency of referral, and the most appropriate initial investigations.
Pattern of Weakness
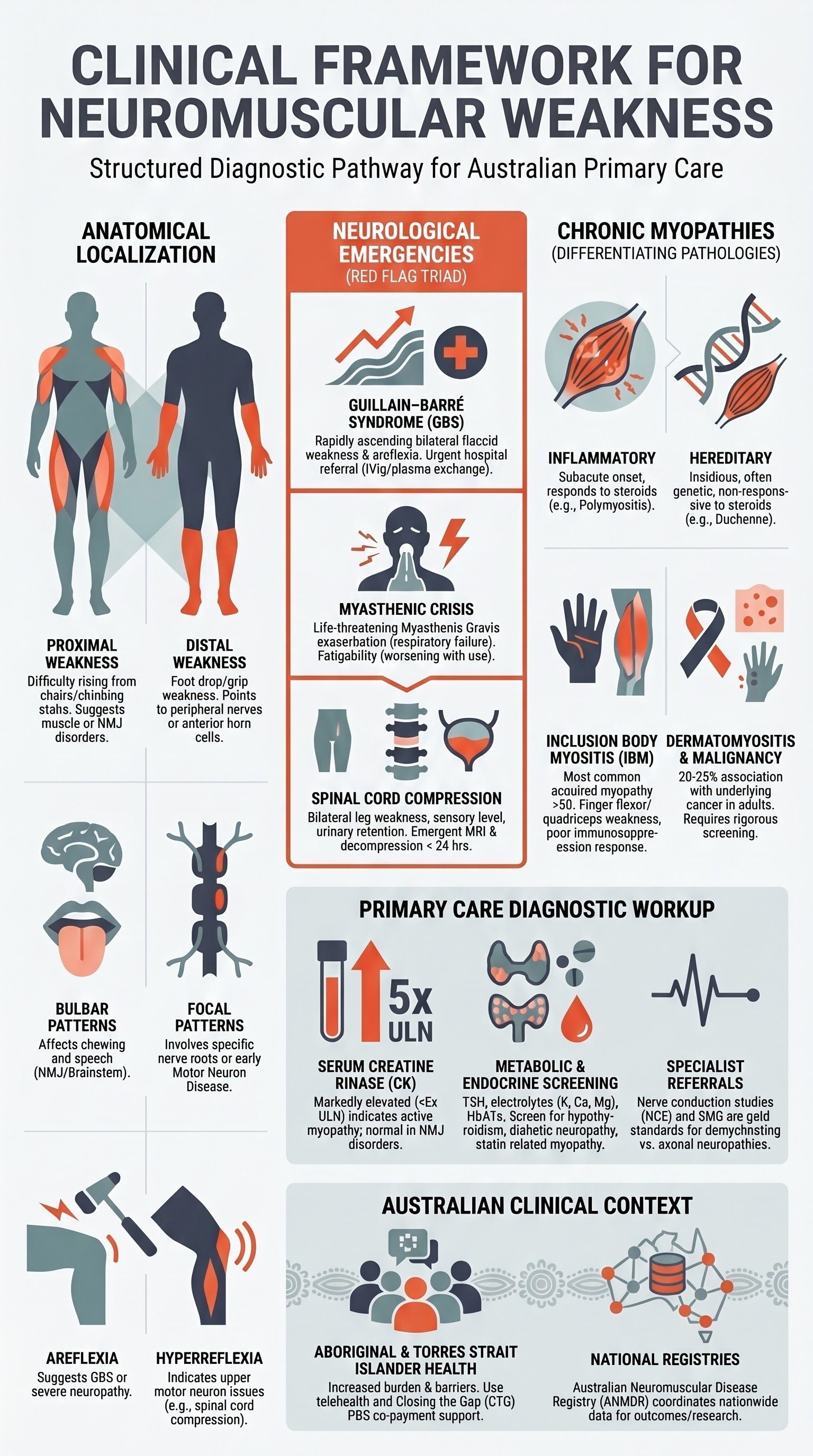
The distribution of weakness is the single most important clinical clue:
| Pattern | Typical Activities Affected | Localisation | Examples |
|---|---|---|---|
| Proximal | Rising from chair, climbing stairs, brushing hair, overhead tasks | Muscle (myopathy) or NMJ | Inflammatory myopathy, statin myopathy, myasthenia gravis, limb-girdle dystrophy |
| Distal | Foot drop, tripping, difficulty with buttons/keys, grip weakness | Peripheral nerve or anterior horn cell | Charcot–Marie–Tooth disease, distal myopathy, motor neuron disease, peripheral neuropathy |
| Focal / Asymmetric | Unilateral hand weakness, single leg dragging | Nerve root, plexus, or anterior horn cell | Radiculopathy, brachial plexopathy, MND (early), mononeuritis multiplex |
| Bulbar | Dysarthria, dysphagia, nasal speech, difficulty chewing | Brainstem, NMJ, or bulbar muscles | Myasthenia gravis, bulbar MND, brainstem stroke |
| Ascending / Symmetric | Bilateral leg weakness progressing to arms over days | Peripheral nerve (demyelinating) | Guillain–Barré syndrome |
Reflexes
Deep tendon reflexes provide critical localising information:
- Areflexia or generalised hyporeflexia — Guillain–Barré syndrome, severe peripheral neuropathy, advanced myopathy (CK often very high)
- Normal reflexes in weak muscles — Consider neuromuscular junction disorder (myasthenia gravis) or early myopathy
- Hyperreflexia with weakness — Suggests upper motor neuron pathology (spinal cord compression, motor neuron disease with UMN features); not a primary muscle or NMJ disorder
- Reflex decrement with fatigability — May be seen in advanced MG but is not a reliable bedside sign
Fatigability
Fatigability — weakness worsening with repeated muscle use and improving with rest — is the hallmark of neuromuscular junction disorders, particularly myasthenia gravis. Bedside tests include:
- Sustained upgaze test: Ask the patient to look upward for 60 seconds; development of ptosis or diplopia is positive.
- Counting test: Ask the patient to count aloud from 1 to 50; progressive nasal speech, hypophonia, or inability to complete suggests bulbar fatigability.
- Arm raise test: Hold arms abducted to 90° for 2 minutes; proximal drop suggests fatigable weakness.
- Ice-pack test: Application of ice over a ptotic eyelid for 2 minutes — improvement of ptosis by ≥2 mm is suggestive of MG (sensitivity ~80%).
Respiratory Red Flags
- Dyspnoea at rest or on minimal exertion
- Orthopnoea or inability to lie flat
- Weak or ineffective cough
- Difficulty swallowing (choking on saliva, nasal regurgitation)
- Nasal speech or hypophonia
- Accessory muscle use, paradoxical abdominal breathing
- Forced vital capacity (FVC) <20 mL/kg or <1 L (if bedside spirometry available)
Urgent Conditions
Three conditions in neuromuscular weakness constitute neurological emergencies: Guillain–Barré syndrome, myasthenic crisis, and spinal cord compression. Rapid recognition and appropriate referral can prevent irreversible disability or death.
Guillain–Barré Syndrome (GBS)
GBS is an acute, immune-mediated polyradiculoneuropathy, most commonly the acute inflammatory demyelinating polyradiculoneuropathy (AIDP) variant in Australia. It typically follows a respiratory or gastrointestinal infection by 1–4 weeks. Campylobacter jejuni is the most common precipitant worldwide.
Clinical Features
- Bilateral, symmetric, ascending flaccid weakness — typically starting in the legs and progressing proximally
- Areflexia or marked hyporeflexia in affected limbs
- Sensory symptoms (paraesthesia, pain) — often present but motor predominates
- Autonomic dysfunction — tachycardia, blood pressure fluctuations, urinary retention, ileus
- Cranial nerve involvement — bilateral facial weakness (50%), bulbar weakness, ophthalmoplegia (Miller Fisher variant)
- Miller Fisher variant — triad of ophthalmoplegia, ataxia, and areflexia with anti-GQ1b antibodies
Brighton Diagnostic Criteria (Modified)
| Feature | Required for Clinical Diagnosis |
|---|---|
| Bilateral limb weakness | Required — progressive over days to 4 weeks |
| Areflexia or hyporeflexia | Required — generalised (in all affected limbs) |
| Monophasic course | Peak within 4 weeks, then plateau or improvement |
| Exclusion of alternative causes | Electrolyte disturbance, myelopathy, botulism, critical illness neuropathy |
| Nerve conduction studies / CSF | Supportive but not essential for clinical diagnosis if pattern is classic |
Investigations
- Nerve conduction studies / EMG: Demyelinating pattern (prolonged distal latencies, conduction block, temporal dispersion, F-wave prolongation) — available at major Australian hospitals; arrange urgently via neurology registrar
- Lumbar puncture: Albuminocytological dissociation (elevated protein with normal cell count) — may be normal in the first week; do not delay treatment if clinical suspicion is high
- Serum anti-ganglioside antibodies: Anti-GM1, anti-GD1a (AMAN variant), anti-GQ1b (Miller Fisher) — available through major pathology services (Sullivan Nicolaides, Douglass Hanly Moir, Melbourne Pathology)
- Respiratory function: Serial FVC measurement — admission to HDU/ICU if FVC <20 mL/kg or declining rapidly
- Autonomic monitoring: Continuous cardiac monitoring, blood pressure, urine output
Treatment
Myasthenic Crisis
Myasthenic crisis is defined as exacerbation of myasthenia gravis causing respiratory failure requiring mechanical ventilation. It occurs in approximately 15–20% of patients with MG at some point in their disease course and carries a mortality rate of approximately 5–10% in modern ICUs.
Triggers
- Infection (most common trigger — respiratory tract infections especially)
- Medication changes — withdrawal of immunosuppression, initiation of aminoglycosides, fluoroquinolones, beta-blockers, magnesium, or neuromuscular blocking agents
- Surgery, particularly thymectomy or any general anaesthesia
- Heat, emotional stress, thyroid dysfunction
- Pregnancy and the postpartum period
Clinical Recognition
- Worsening bulbar weakness — dysphagia, nasal speech, inability to manage secretions
- Progressive respiratory weakness — dyspnoea, shallow breathing, declining FVC
- Generalised limb weakness
- Distinguish from cholinergic crisis (excess anticholinesterase medication): cholinergic crisis features miosis, bradycardia, sweating, salivation, GI cramping — these features are absent in myasthenic crisis
Acute Treatment
- Intubate early if FVC <15 mL/kg, declining rapidly, or bulbar failure with aspiration risk
- Avoid succinylammonium for rapid-sequence intubation — use rocuronium or propofol/ketamine with caution; prolonged paralysis may occur
- Temporarily discontinue anticholinesterase agents (pyridostigmine) during acute crisis — they increase secretions and may worsen respiratory management
- Screen for and treat triggers (infection, thyroid dysfunction, medication interactions)
Spinal Cord Compression
Spinal cord compression is a neurological emergency requiring rapid recognition and decompression. It may arise from metastatic disease (most common in Australia: lung, breast, prostate, kidney, colorectal), epidural abscess, haematoma, or acute disc herniation.
Clinical Features
- Motor: Bilateral leg weakness (paraparesis or paraplegia), initially flaccid (spinal shock) then spastic
- Sensory: Sensory level — band-like dermatomal distribution, posterior column loss (proprioception, vibration)
- Sphincter: Urinary retention (often the earliest autonomic sign), constipation, bowel incontinence
- Pain: Localised spinal pain, often worse when lying down (nocturnal exacerbation in metastatic disease), radicular pain
- Reflexes: Initially areflexic (spinal shock), then hyperreflexic with extensor plantar responses (upper motor neuron signs)
Immediate Management
- Emergent MRI of the whole spine — must be performed within 24 hours of suspected cord compression
- Dexamethasone 10 mg IV stat, then 4 mg IV/PO QID — reduces vasogenic oedema; taper over 7–14 days
- Surgical decompression — within 24 hours if operable; consult neurosurgery or spinal surgery urgently
- Radiation oncology — for inoperable metastatic disease; emergency radiation therapy within 24–48 hours
- Bladder management — indwelling catheter for urinary retention; monitor input/output
Chronic Myopathies
Chronic myopathies present with insidious progressive weakness, typically proximal, over weeks to months to years. The key clinical question in primary care is whether the presentation suggests an inflammatory myopathy (potentially treatable with immunosuppression) or a hereditary myopathy (requiring genetic diagnosis and supportive management).
Inflammatory vs Hereditary Myopathy — Clinical Clues
| Feature | Inflammatory Myopathy | Hereditary Myopathy |
|---|---|---|
| Onset | Subacute (weeks to months) | Insidious (months to years); may present in childhood |
| Age at onset | Any age; dermatomyositis bimodal (paediatric + adult peaks); inclusion body myositis >50 years | Childhood/adolescence for Duchenne/Becker; variable for limb-girdle; 20–40 years for FSHD, myotonic dystrophy |
| Family history | Usually negative | Often positive — autosomal dominant (FSHD, myotonic dystrophy) or recessive (limb-girdle); X-linked (Duchenne, Becker) |
| CK level | Elevated 5–50× upper limit of normal (ULN); may be normal in IBM | Variable — markedly elevated in Duchenne/Becker (50–100× ULN); mildly elevated in FSHD; may be normal in some limb-girdle types |
| Skin changes | Present in dermatomyositis — heliotrope rash, Gottron's papules, V-sign, shawl sign, mechanic's hands | Absent |
| Myotonia | Absent | Present in myotonic dystrophy — inability to relax grip, percussion myotonia of thenar eminence |
| Pattern of weakness | Symmetric proximal; IBM: distal (finger flexors) + proximal + quadriceps | Facioscapulohumeral: face, scapular stabilisers, biceps; Limb-girdle: shoulder and hip girdle; Myotonic: distal (hands, feet) + facial |
| Response to steroids | Polymyositis and dermatomyositis — usually responsive; IBM — poor or absent response | Not responsive |
| Extramuscular features | Interstitial lung disease (anti-Jo-1), malignancy (dermatomyositis — ovarian, lung, GI, lymphoma), dysphagia, cardiac involvement | Cardiac involvement (myotonic dystrophy — conduction defects, cardiomyopathy); cataracts (myotonic dystrophy); respiratory muscle weakness |
Inflammatory Myopathies — Detailed Approach
Polymyositis & Dermatomyositis
Dermatomyositis (DM) is characterised by proximal muscle weakness plus characteristic cutaneous features and carries a significant association with underlying malignancy (estimated 20–25% in adults). Polymyositis (PM) presents with isolated proximal weakness without skin changes. Both are more common in women.
Malignancy screening in adult dermatomyositis:
- Age-appropriate cancer screening (colonoscopy, mammography, cervical screening, prostate-specific antigen)
- CT chest/abdomen/pelvis with contrast
- Pelvic ultrasound (ovarian malignancy screening — particularly important in women)
- PET-CT if available and clinical suspicion remains high
- Anti-TIF1-γ and anti-NXP2 antibodies are associated with cancer-associated DM
- Screening should occur within 3 months of DM diagnosis and continue for 3–5 years
Inclusion Body Myositis (IBM)
IBM is the most common acquired myopathy in patients over 50 years and is often misdiagnosed as polymyositis. Key distinguishing features include:
- Slowly progressive course (over years, not weeks)
- Predominant involvement of finger flexors (difficulty opening jars, turning keys) and quadriceps (tendency to fall)
- CK may be normal or only mildly elevated (2–5× ULN)
- Poor or no response to immunosuppressive therapy — this is a critical distinction from PM/DM
- Muscle biopsy shows endomysial inflammation with rimmed vacuoles (pathognomonic)
- Associated with anti-cN1A (NT5C1A) antibodies
Initial Treatment — Inflammatory Myopathies (PM/DM, not IBM)
Hereditary Myopathies — Overview
Hereditary myopathies are a genetically heterogeneous group of disorders. Referral to a neuromuscular specialist and clinical genetics service is essential for definitive diagnosis, genetic counselling, and family planning advice.
| Condition | Inheritance | Key Clinical Features | Gene / Genetic Test | Key Monitoring |
|---|---|---|---|---|
| Duchenne Muscular Dystrophy (DMD) | X-linked recessive | Onset 2–5 years; proximal weakness, Gowers' sign, calf pseudohypertrophy, wheelchair by 12 years, respiratory/cardiac failure by 20s | DMD gene (dystrophin); MLPA, sequencing | CK (markedly elevated), cardiac MRI, respiratory function, corticosteroid therapy from age 4–6 |
| Facioscapulohumeral Dystrophy (FSHD) | Autosomal dominant | Facial weakness (inability to whistle/close eyes), scapular winging, foot drop; may be asymmetric; CK normal or mildly elevated | D4Z4 repeat contraction (FSHD1); SMCHD1 variants (FSHD2); molecular testing via Southern blot / optical genome mapping | Audiometry, retinal examination (Coats disease in paediatric onset), respiratory function, cardiac assessment |
| Myotonic Dystrophy Type 1 (DM1) | Autosomal dominant (CTG expansion) | Distal weakness, grip myotonia, facial weakness (hatchet face), cataracts, cardiac conduction defects, excessive daytime somnolence, insulin resistance | DMPK gene CTG repeat; blood-based PCR / Southern blot | ECG (annual), echocardiogram, HbA1c, sleep studies, ophthalmology review; anaesthetic risk assessment (malignant hyperthermia-like reactions) |
| Limb-Girdle Muscular Dystrophy (LGMD) | AD or AR (multiple subtypes) | Proximal weakness of shoulder and hip girdles; variable age of onset and severity; CK typically elevated | Multiple genes (sarcoglycans, calpain-3, dysferlin, etc.); next-generation sequencing panel | Cardiac and respiratory monitoring; genetic counselling |
Investigations
Initial Investigations — Primary Care
The following investigations can be ordered in primary care to narrow the differential and guide referral:
Specialist Investigations — Referral-Based
Special Populations
Pregnancy
Paediatrics
Elderly
Renal Impairment
Hepatic Impairment
Immunocompromised
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Leonhard SE, Mandarakas MR, Gondim FAA, et al. Diagnosis and management of Guillain–Barré syndrome in ten steps. Nat Rev Neurol. 2019;15(11):671–683.
- 2. Bershad EM, Feen ES, Suarez JI. Myasthenia crisis. Curr Treat Options Neurol. 2011;13(1):67–79.
- 3. Lundberg IE, Tjärnlund A, Bottai M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis. 2017;76(12):1955–1964.
- 4. Dimachkie MM, Barohn RJ. Inclusion body myositis. Curr Neurol Neurosci Rep. 2013;13(1):321.
- 5. Gilhus NE, Tzartos S, Evoli A, Palace J, Burns TM, Verschuuren JJGM. Myasthenia gravis. Nat Rev Dis Primers. 2019;5(1):30.
- 6. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander Health Performance Framework. Canberra: AIHW; 2023. Available at: aihw.gov.au.
- 7. Wandrag L, Garg S, Grzechnik E, et al. The Australian Neuromuscular Disease Registry: a national approach to improving outcomes. J Neuromuscul Dis. 2021;8(s2):S323–S331.
- 8. Hughes RAC, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain–Barré syndrome. Cochrane Database Syst Rev. 2014;(9):CD002063.
- 9. Dalakas MC. Inflammatory myopathies: management of steroid resistance. Curr Opin Neurol. 2011;24(5):457–462.
- 10. Narayanaswami P, Sanders DB, Wolfe G, et al. International consensus guidance for management of myasthenia gravis: 2020 update. Neurology. 2021;96(3):114–122.
- 11. Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9(1):77–93.
- 12. Tawil R, Kissel JT, Heatwole C, et al. Evidence-based guideline summary: evaluation, diagnosis, and management of facioscapulohumeral muscular dystrophy. Neurology. 2015;85(4):357–364.
- 13. Gourie-Devi M. Guillain–Barré syndrome in pregnancy: a review. Ann Indian Acad Neurol. 2018;21(2):109–115.
- 14. National Health and Medical Research Council (NHMRC). National Statement on Ethical Conduct in Human Research. Canberra: NHMRC; 2023. [Relevant to genetic testing consent frameworks].
- 15. Royal Australian College of General Practitioners (RACGP). Management of type 2 diabetes: A handbook for general practice. Melbourne: RACGP; 2020. [Chapter on diabetic neuropathy screening].