📋 Key Information Summary
- B cell diversity enables the humoral immune system to recognise an estimated 1011 distinct antigenic epitopes from approximately 25 000 human genes.
- V(D)J recombination — mediated by RAG-1/RAG-2 recombinases — randomly assembles variable (V), diversity (D), and joining (J) gene segments in the heavy-chain locus and V–J segments in each light-chain locus during B cell development in the bone marrow.
- Combinatorial diversity alone generates >6 000 heavy-chain and >170 light-chain combinations; junctional diversity (P-nucleotides, N-nucleotide addition by TdT, and exonuclease trimming) multiplies this figure by orders of magnitude.
- Failure of V(D)J recombination (e.g. RAG1/RAG2, Artemis, DNA-PKcs mutations) causes severe combined immunodeficiency (SCID), typically presenting in infancy with recurrent sinopulmonary and opportunistic infections.
- Somatic hypermutation (SHM) introduces point mutations in immunoglobulin variable regions at a rate ~106× the background genomic mutation rate, driven by activation-induced cytidine deaminase (AID).
- Affinity maturation is the iterative process of clonal selection within germinal centres whereby B cells with higher-affinity BCRs are preferentially expanded, progressively increasing serum antibody affinity over weeks to months.
- Class-switch recombination (CSR), also AID-dependent, changes the constant-region isotype (IgM → IgG, IgA, or IgE) without altering antigen specificity.
- Hyper-IgM syndromes (AID deficiency, UNG deficiency, CD40L deficiency) impair CSR and/or SHM, causing susceptibility to encapsulated bacteria and Pneumocystis jirovecii.
- Defective SHM and aberrant AID activity are implicated in B cell lymphomagenesis, particularly diffuse large B cell lymphoma (DLBCL) and Burkitt lymphoma.
- In Australia, genetic testing for SCID and hyper-IgM syndromes is available through National Association of Testing Authorities (NATA)-accredited laboratories and the Australian Genomics Health Alliance; newborn screening for SCID via TREC assay is now operational in all states and territories.
- Aboriginal and Torres Strait Islander children experience higher rates of invasive pneumococcal disease and suppurative otitis media — conditions exacerbated by impaired antibody maturation — underscoring the importance of early recognition of humoral immunodeficiency.
- Understanding the molecular mechanisms of B cell diversity is essential for interpreting flow-cytometric B cell subset analysis (naïve, memory, switched memory, plasmablasts) and diagnosing primary immunodeficiency disorders in Australian clinical practice.
Introduction & Australian Context
The adaptive humoral immune system depends on the generation of a vast repertoire of B cell receptors (BCRs) — membrane-bound immunoglobulins — capable of recognising virtually any molecular structure encountered in a lifetime. This extraordinary diversity is achieved through a series of molecular mechanisms operating at distinct stages of B cell development and antigen-driven maturation. Disruption of any step in this cascade results in clinically significant primary immunodeficiency, autoimmunity, or lymphoid malignancy.
B cell development begins in the foetal liver and transitions to the bone marrow by mid-gestation, continuing throughout life. In Australia, the estimated prevalence of primary immunodeficiency disorders (PIDs) is approximately 1 in 1 200 to 1 in 2 500 live births, with humoral defects (including common variable immunodeficiency [CVID], X-linked agammaglobulinaemia [XLA], and hyper-IgM syndromes) accounting for >50 % of diagnosed cases (AIHW, 2023). The Australian Paediatric Surveillance Unit (APSU) and the Australasian Society of Clinical Immunology and Allergy (ASCIA) PID Registry continue to collect national data on these conditions.
This article reviews the four principal mechanisms generating B cell diversity — V(D)J recombination, junctional diversity, somatic hypermutation, and affinity maturation — with emphasis on their molecular biology, clinical consequences of disruption, and relevance to Australian diagnostic and therapeutic practice.
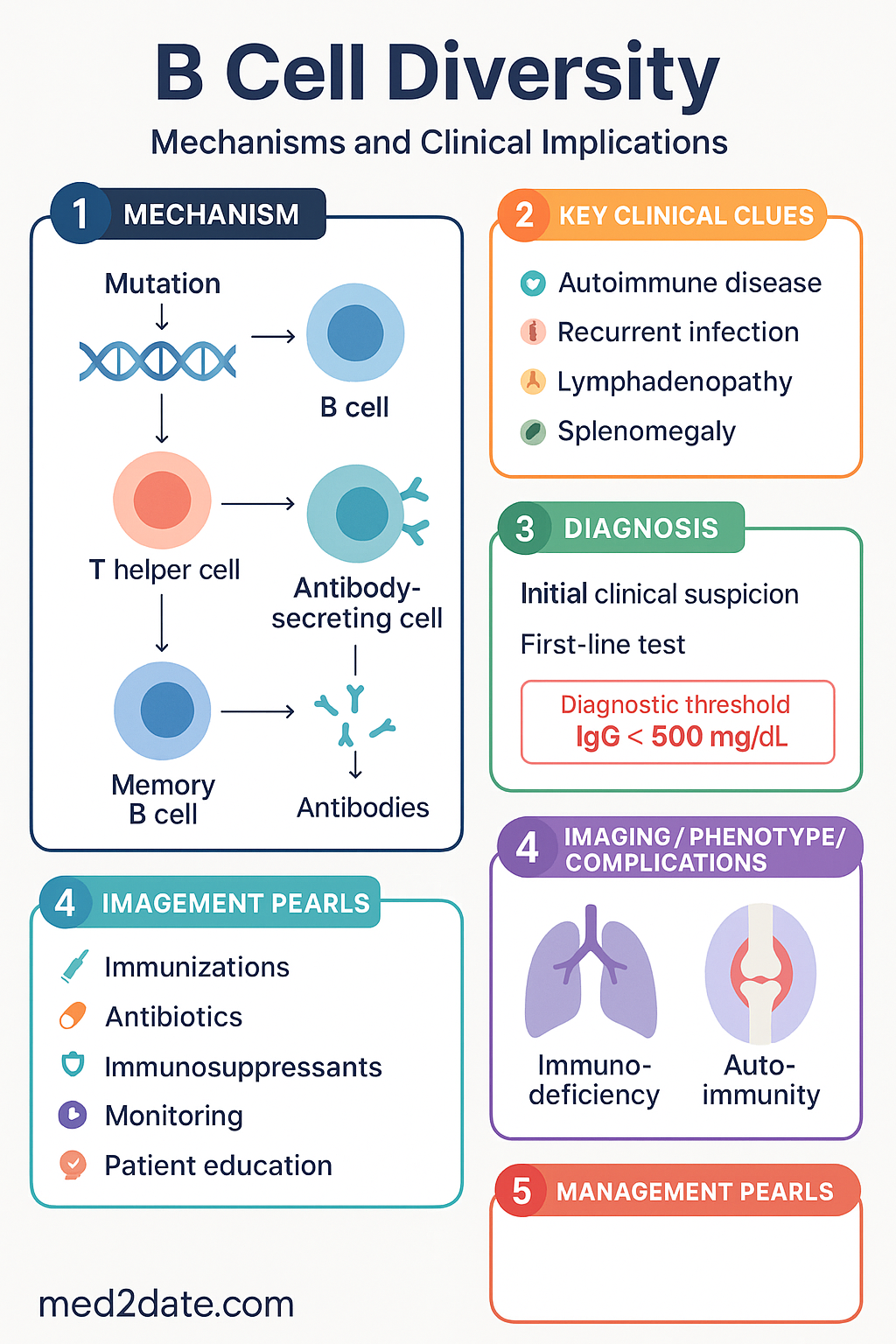
V(D)J Recombination
Molecular Mechanism
V(D)J recombination is the primary somatic rearrangement event that assembles a functional immunoglobulin variable-region exon from germline gene segments. It occurs exclusively in developing B cells (and T cells for TCR genes) within the bone marrow, prior to antigen encounter.
The immunoglobulin heavy-chain locus (IGH, chromosome 14q32) contains approximately 45 functional VH segments, 23 DH segments, and 6 JH segments. Each light-chain locus — IGK (2p11) and IGL (22q11) — contains V and J segments only (no D segments). Recombination proceeds in two ordered steps:
- Heavy-chain rearrangement: DH→JH first, followed by VH→DJH. A productive rearrangement (in-frame, no stop codons) produces a μ heavy chain and signals allelic exclusion at the second IGH allele.
- RAG-1 / RAG-2 Recombinases
Recombination-activating genes 1 and 2 (RAG1, RAG2) form a heterotetrameric complex that introduces double-strand DNA breaks at recombination signal sequences (RSSs) flanking each V, D, and J segment. RSSs consist of a conserved heptamer, a nonamer, and a spacer of either 12 bp or 23 bp; efficient recombination requires one 12-RSS and one 23-RSS (the 12/23 rule). RAG deficiency results in a complete block in both B and T cell development — a form of T−B− NK+ SCID.
Clinical significance: RAG1/RAG2 mutations account for approximately 10–15 % of all SCID cases in Australia. Newborn screening using the T-cell receptor excision circle (TREC) assay detects low or absent TRECs, enabling pre-symptomatic diagnosis and referral for haematopoietic stem cell transplantation (HSCT).Combinatorial Diversity Calculation
Locus Functional V Functional D Functional J V×D×J or V×J Combinations IGH (heavy chain) ~45 ~23 6 ~6 210 IGK (κ light chain) ~40 — 5 ~200 IGL (λ light chain) ~30 — 4 ~120 Combinatorial diversity of heavy + light chains (6 210 × 320) yields approximately 2 × 106 unique BCRs before junctional diversity is considered.
Junctional Diversity
Mechanisms
Junctional diversity dramatically increases receptor diversity beyond combinatorial rearrangement alone. Three enzymatic processes modify the junctions (CDR3 regions) between joined gene segments:
- P-nucleotides (palindromic nucleotides): Asymmetric hairpin opening by Artemis (an endonuclease activated by DNA-PKcs) creates short single-stranded overhangs complementary to the coding end. These are filled in by DNA polymerase, adding a few palindromic nucleotides.
- N-nucleotide addition: Terminal deoxynucleotidyl transferase (TdT), expressed primarily in developing lymphocytes, adds non-templated (random) nucleotides at the D–J and V–D junctions of heavy chains. N-addition is minimal or absent in light chains and in foetal/neonatal B cells, explaining the more restricted neonatal repertoire.
- Exonuclease trimming: Unregulated 5′→3′ and 3′→5′ exonuclease activity removes nucleotides from coding ends before ligation, further diversifying CDR3 length and sequence.
Magnitude of Diversity
Junctional diversity, concentrated in CDR3 — the most hypervariable and antigen-contacting region of the antibody — increases theoretical repertoire size to >1011 unique specificities. CDR3 length variation (heavy-chain CDR3: 3–25 amino acids) is a major contributor to fine specificity differences between antibodies recognising the same antigen.
Clinical Correlates of Defective Junctional Diversity
Somatic Hypermutation (SHM)
Mechanism
Somatic hypermutation is an antigen-driven, post-germinal-centre diversification mechanism that introduces point mutations into rearranged immunoglobulin variable-region genes at a rate of approximately 10−3 mutations per base pair per cell division — roughly 106 times the background somatic mutation rate. SHM is confined to a ~2 kb region spanning the rearranged V(D)J exon and its flanking intron, targeting hotspot motifs (WRCY / RGYW, where W = A/T, R = A/G, Y = C/T).
Activation-Induced Cytidine Deaminase (AID)
AID (encoded by AICDA, chromosome 12p13) is the essential enzyme initiating both SHM and class-switch recombination. It deaminates cytosine to uracil in single-stranded DNA exposed during transcription, creating U:G mismatches. These mismatches are processed by three downstream pathways:
- Replication over uracil → C→T transitions (direct pathway).
- Uracil-DNA glycosylase (UNG) excision → abasic site → error-prone repair → transitions and transversions.
- Mismatch repair (MSH2/MSH6) → error-prone polymerase recruitment (Rev1, Polη) → broader mutation spectrum including mutations at A:T base pairs.
AID Deficiency (Hyper-IgM Syndrome Type 2)
AID deficiency (autosomal recessive, AICDA mutations) abolishes both SHM and CSR while preserving V(D)J recombination. Patients present with normal or elevated IgM but absent IgG, IgA, and IgE, combined with loss of somatic mutations in Ig genes. Clinical features include recurrent sinopulmonary infections (encapsulated organisms), lymphoid hyperplasia, and susceptibility to Giardia lamblia and enteroviral infections.
Diagnosis in Australia
Pharmacological Modulation of SHM
Affinity Maturation
Germinal Centre Biology
Affinity maturation is the progressive increase in the binding affinity of serum antibodies following repeated antigen exposure. It occurs within germinal centres (GCs) — specialised microanatomical structures that form in secondary lymphoid organs (lymph nodes, spleen, Peyer's patches) 7–10 days after initial antigen encounter.
The germinal centre is organised into two functionally distinct zones:
- Dark zone: Contains rapidly dividing centroblasts that undergo SHM of their BCR variable-region genes. Each division introduces 1–3 mutations per V-region exon.
- Light zone: Contains centrocytes that have ceased dividing and express mutated BCRs on their surface. Centrocytes compete for limiting antigen displayed as immune complexes on follicular dendritic cells (FDCs). Only centrocytes with the highest-affinity BCRs receive T cell help from follicular helper T cells (TFH) via CD40L–CD40 interaction, ICOS–ICOSL, and IL-21 signalling.
Centrocytes that receive adequate TFH help (survival signal) differentiate into either memory B cells or long-lived antibody-secreting plasma cells. Those that fail to capture antigen or receive insufficient help undergo apoptosis — a process termed clonal selection within the germinal centre.
Iterative Cycling
Selected centrocytes may re-enter the dark zone for additional rounds of SHM and selection, creating an iterative cycle (cyclic re-entry model) that progressively refines antibody affinity over weeks to months. Serial serum sampling during primary immunisation demonstrates a measurable increase in antibody affinity (measured by avidity ELISA or surface plasmon resonance) over 4–8 weeks following the first dose, with further increases after booster doses.
Quantifying Affinity Maturation
| Parameter | Primary Response | Secondary Response | Clinical Application |
|---|---|---|---|
| Antibody isotype | Predominantly IgM | Predominantly IgG (class-switched) | IgG avidity testing for CMV, rubella, measles timing |
| Affinity (KD) | Low (10−6–10−7 M) | High (10−9–10−11 M) | Vaccine efficacy assessment; defining protective thresholds |
| SHM load (mutations/V-gene) | Germline (0 mutations) | 5–30+ mutations per V-gene | CVID classification; lymphoma clonality analysis |
| Time to peak | 7–14 days | 3–7 days (memory recall) | Vaccination timing in immunocompromised hosts |
Clinical Implications in Australia
Impaired affinity maturation is a hallmark of CVID and specific antibody deficiency (SAD). Australian immunologists assess vaccine responses by measuring pre- and post-vaccination (4–8 weeks) titres and serotype-specific IgG for pneumococcal polysaccharide (Pneumovax® 23) or conjugate (Prevenar 13®) vaccines, with additional avidity testing where indicated. Failure to mount a ≥2-fold rise in protective titres for ≥50 % of serotypes tested indicates a need for immunoglobulin replacement therapy.
Pathophysiology — Integrated View
B cell diversity is generated through sequential, overlapping mechanisms that collectively produce a repertoire of >1011 unique specificities from a limited germline genome:
Defects at any stage — RAG (SCID), Artemis/DNA-PKcs (junctional), AID/UNG (SHM/CSR), CD40L/CD40 (GC formation) — produce distinct clinical phenotypes collectively classified as primary B cell or combined immunodeficiency disorders.
Risk Stratification & Severity Scoring
Disorders of B cell diversity can be stratified by severity based on the affected mechanism, residual immune function, and timing of presentation:
Management & Therapeutic Approaches
Immunoglobulin Replacement Therapy
The cornerstone of management for hypogammaglobulinaemia secondary to impaired B cell diversity (CVID, hyper-IgM, XLA). Replacement provides functional antibodies the patient cannot produce due to defective affinity maturation and class switching.
Prophylactic Antimicrobials
Haematopoietic Stem Cell Transplantation (HSCT)
HSCT is the definitive treatment for SCID and is curative when performed early (ideally before 3.5 months of age and before onset of severe infection). In Australia, HSCT for SCID is performed at:
- The Children's Hospital at Westmead, Sydney
- Royal Children's Hospital, Melbourne
- Queensland Children's Hospital, Brisbane
- Princess Margaret Hospital / Perth Children's Hospital, Perth
- Women's and Children's Hospital, Adelaide
Monitoring
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Tonegawa S. Somatic generation of antibody diversity. Nature. 1983;302(5909):575–581. doi:10.1038/302575a0
- 2. Schatz DG, Ji Y. Recombination centres and the orchestration of V(D)J recombination. Nature Reviews Immunology. 2011;11(4):251–263. doi:10.1038/nri2941
- 3. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102(5):553–563. doi:10.1016/S0092-8674(00)00078-7
- 4. Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annual Review of Biochemistry. 2007;76:1–22. doi:10.1146/annurev.biochem.76.061705.090740
- 5. Victora GD, Nussenzweig MC. Germinal centers. Annual Review of Immunology. 2022;40:413–442. doi:10.1146/annurev-immunol-120419-022408
- 6. Tangye SG, Al-Herz W, Bousfiha A, et al. Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee. Journal of Clinical Immunology. 2022;42(7):1473–1507. doi:10.1007/s10875-022-01289-3
- 7. Australasian Society of Clinical Immunology and Allergy (ASCIA). Primary immunodeficiency diseases (PIDs) — position statements and clinical resources. ASCIA; 2024. https://www.allergy.org.au/patients/primary-immunodeficiency-diseases
- 8. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander health performance framework: immunisation and infectious diseases. AIHW; 2023. https://www.aihw.gov.au/reports/indigenous-australians/indigenous-health-performance-framework
- 9. Kwan A, Abraham RS, Currier R, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. 2014;312(7):729–738. doi:10.1001/jama.2014.9132
- 10. Chapel H, Lucas M, Lee M, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112(2):277–286. doi:10.1182/blood-2007-11-124545
- 11. Revy P, Muto T, Levy Y, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell. 2000;102(5):565–575. doi:10.1016/S0092-8674(00)00079-9
- 12. Bobby Gaspar H, Thrasher AJ. Gene therapy for severe combined immunodeficiency due to adenosine deaminase deficiency. Immunology and Allergy Clinics of North America. 2010;30(2):203–219. doi:10.1016/j.iac.2010.01.003
- 13. Central Australian Rural Practitioners Association (CARPA). CARPA Standard Treatment Manual. 8th ed. Alice Springs: CARPA; 2022.
- 14. National Health and Medical Research Council (NHMRC). The Australian Immunisation Handbook. Australian Government Department of Health; 2024 (updated). https://immunisationhandbook.health.gov.au