📋 Key Information Summary
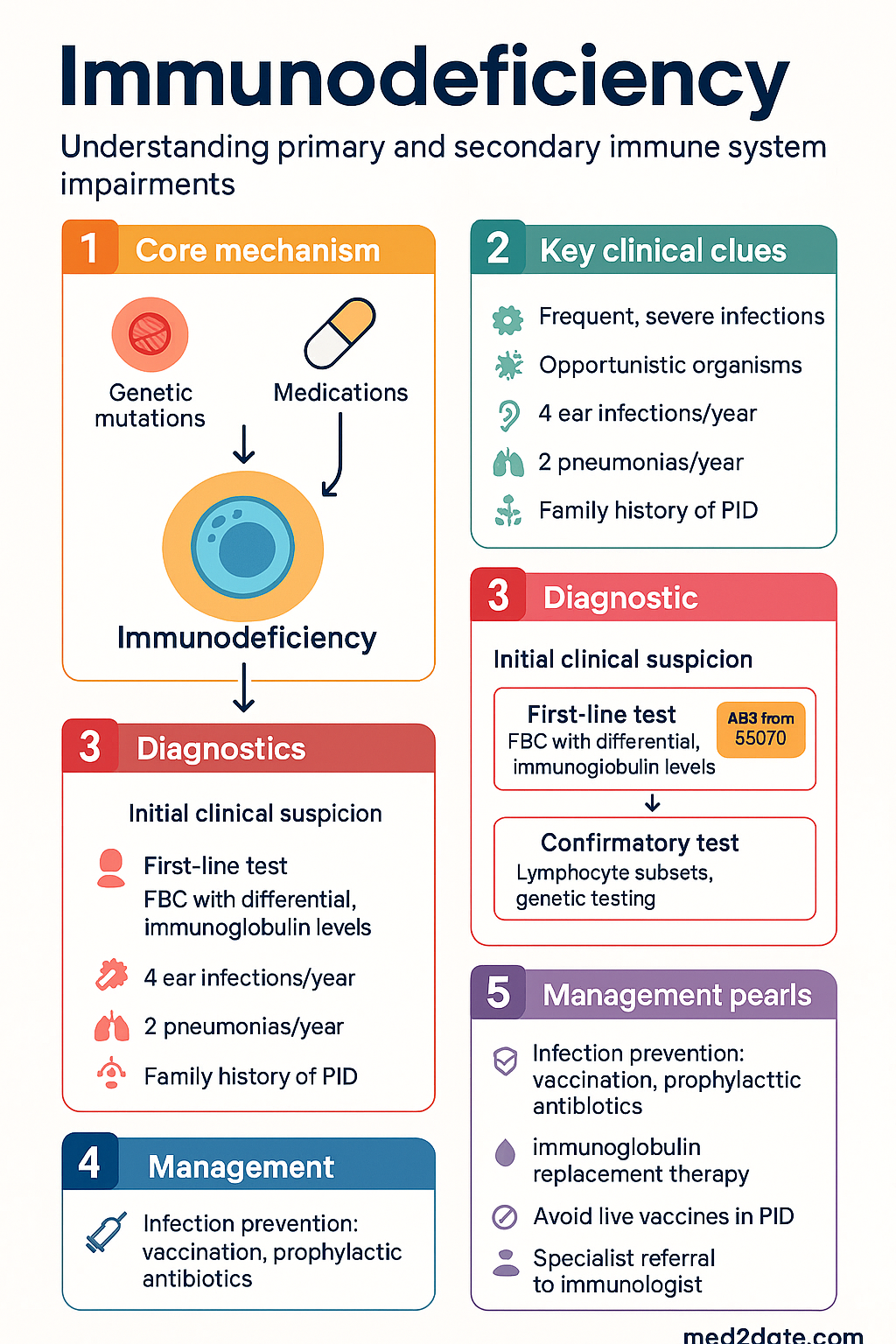
- Immunodeficiency is classified as primary (intrinsic immune defect, often genetic) or secondary (acquired due to disease, medication, or environmental factors).
- Secondary immunodeficiency is far more common in Australian primary care; primary immunodeficiency (PID) affects an estimated 1 in 1,200–2,000 Australians, though many remain undiagnosed.
- Suspect immunodeficiency when infections are unusually frequent, severe, atypical, or caused by opportunistic organisms.
- Red-flag features include: ≥4 ear infections/year, ≥2 pneumonias/year, prolonged IV antibiotics required, failure to thrive, deep-organ abscesses, or family history of PID.
- Initial investigations include FBC with differential, immunoglobulin levels (IgG, IgA, IgM, IgE), complement (CH50/C3/C4), lymphocyte subsets, and basic lymphocyte function (e.g., tetanus/diphtheria serology).
- MBS items exist for immunoglobulin quantification (MBS item 65070) and lymphocyte subsets (MBS item 65073); genetic/genomic testing is increasingly accessible through specialised centres.
- Management centres on infection prevention (vaccination, prophylactic antibiotics, immunoglobulin replacement), definitive therapy (haematopoietic stem-cell transplant, gene therapy for severe forms), and treatment of complications (autoimmunity, granulomas, lymphoma).
- Immunoglobulin replacement (IVIG or SCIG) is the cornerstone therapy for antibody-deficient patients; PBS Authority Required for ≥2 indications.
- Live vaccines (MMR, varicella, BCG, oral polio, yellow fever) are contraindicated in most PID; assess with an immunologist before live-vaccine administration in any suspected immunodeficiency.
- Glucocorticoids ≥20 mg/day prednisolone for ≥14 days, anti-TNF agents, rituximab, and cytotoxic chemotherapy are the most common causes of secondary immunodeficiency in Australian practice.
- Refer to a clinical immunologist/allergist for any patient with suspected PID; early referral improves outcomes and reduces organ damage.
- Aboriginal and Torres Strait Islander peoples experience a disproportionate burden of secondary immunodeficiency related to chronic disease, rheumatic heart disease management, and remote access barriers to immunoglobulin supply.
- Transition of care for adolescents with PID from paediatric to adult services should be actively managed using a structured transition plan.
Introduction & Australian Epidemiology
Immunodeficiency encompasses states in which the immune system's ability to fight infections and malignancies is compromised. The disorders are broadly classified as primary (intrinsic genetic or developmental defects of the immune system) or secondary (acquired through disease processes, medications, or environmental exposure). Immunodeficiency Australia-wide represents a significant and growing clinical burden, driven by an ageing population, expanding use of immunosuppressive biologics, and improved recognition of previously undiagnosed primary immunodeficiencies (PIDs).
Over 480 distinct primary immunodeficiency disorders are now catalogued by the International Union of Immunological Societies (IUIS), ranging from severe combined immunodeficiency (SCID) presenting in neonates to common variable immunodeficiency (CVID) diagnosed in adulthood. The Australian Immunodeficiency Registry and international prevalence data suggest approximately 1 in 1,200–2,000 Australians are affected by a PID, yet many individuals remain undiagnosed for years, accumulating preventable organ damage.
Secondary immunodeficiency is vastly more prevalent. Immunosuppressive therapy with glucocorticoids, calcineurin inhibitors, anti-TNF biologics, anti-CD20 agents (rituximab), and cytotoxic chemotherapy affects hundreds of thousands of Australians being treated for autoimmune disease, organ transplantation, and malignancy. The COVID-19 pandemic further highlighted the clinical significance of secondary immunodeficiency, with iatrogenic immunosuppression identified as a major risk factor for severe outcomes.
This guideline provides an evidence-based framework for the recognition, investigation, and management of both primary and secondary immunodeficiency in the Australian healthcare context, including considerations for PBS-listed therapies, MBS investigations, vaccination strategies, and health equity for Aboriginal and Torres Strait Islander peoples.
Primary vs Secondary Immunodeficiency
Understanding the distinction between primary and secondary immunodeficiency is fundamental to appropriate investigation and management. While both share the endpoint of impaired host defence, they differ markedly in aetiology, population affected, diagnostic approach, and therapeutic strategy.
| Feature | Primary Immunodeficiency (PID) | Secondary Immunodeficiency (SID) |
|---|---|---|
| Aetiology | Inborn genetic mutations affecting immune cell development, function, or regulation | Acquired: medications (glucocorticoids, biologics, cytotoxics), infection (HIV), malnutrition, malignancy, nephrotic syndrome, burns |
| Age of onset | Variable — severe forms (SCID) present in infancy; others (CVID, specific antibody deficiency) present in adolescence/adulthood | Any age; predominantly adults on immunosuppressive therapy |
| Prevalence | ~1:1,200–2,000 (estimates; significant under-diagnosis) | Very common — hundreds of thousands of Australians affected |
| Family history | Often positive (autosomal recessive, X-linked, or autosomal dominant patterns) | Not typically relevant |
| Infection pattern | Often severe, recurrent, caused by opportunistic organisms; may involve atypical sites (liver abscess, brain abscess) | Proportionate to degree/type of immunosuppression; common pathogens predominate (e.g., encapsulated organisms in hypogammaglobulinaemia) |
| Genetic testing | Frequently diagnostic (next-generation sequencing panels, whole-exome/genome sequencing) | Not applicable |
| Definitive therapy | HSCT, gene therapy for severe forms; immunoglobulin replacement for antibody deficiencies | Identify and treat underlying cause; reduce/withdraw immunosuppression if safe; prophylactic antimicrobials |
| Reversibility | Generally permanent (except transient hypogammaglobulinaemia of infancy) | Often reversible if causative factor withdrawn |
Primary Immunodeficiency — IUIS Classification (Simplified)
| Category | Key Examples | Typical Defect |
|---|---|---|
| Combined immunodeficiencies | SCID, Omenn syndrome, ZAP-70 deficiency | T-cell ± B-cell development |
| Predominantly antibody deficiencies | CVID, XLA (Bruton's), specific antibody deficiency, transient hypogammaglobulinaemia of infancy | B-cell maturation, class-switch recombination |
| Combined immunodeficiencies with syndromic features | Wiskott–Aldrich syndrome, DiGeorge syndrome (22q11.2 deletion), Ataxia-telangiectasia | Multi-system (thymic, skeletal, neurological) |
| Phagocyte defects | Chronic granulomatous disease (CGD), severe congenital neutropenia, leukocyte adhesion deficiency | Oxidative burst, adhesion, chemotaxis |
| Complement deficiencies | C1q, C2, C3, C4, terminal pathway deficiencies | Classical, lectin, or terminal complement pathway |
| Immune dysregulation disorders | IPEX, APECED, CTLA-4 haploinsufficiency, LRBA deficiency | Regulatory T-cell function, immune tolerance |
| Inborn errors of immunity affecting intrinsic/innate immunity | TLR pathway defects, STAT1/STAT3 GOF/LOF | Pattern recognition, interferon signalling |
Common Causes of Secondary Immunodeficiency in Australia
Clinical Features
The clinical presentation of immunodeficiency is heterogeneous, ranging from neonatal emergencies (SCID) to insidious-onset recurrent infections in adults (CVID). A high index of suspicion is required, as the presenting features often mimic common infectious or inflammatory conditions.
Warning Signs — The Jeffrey Modell Foundation "10 Warning Signs"
- ≥4 new ear infections within 12 months
- ≥2 serious sinus infections within 12 months
- ≥2 months on antibiotics with little effect
- ≥2 pneumonias within 12 months
- Failure to thrive or poor weight gain in infants
- Recurrent deep skin or organ abscesses
- Persistent oral thrush or cutaneous fungal infection beyond infancy
- Need for IV antibiotics to clear infections
- ≥2 deep-seated infections (meningitis, osteomyelitis, septicaemia)
- Family history of primary immunodeficiency
Clinical Features by Immunodeficiency Type
Infections Suggestive of Specific Immune Defects
| Immune Component Affected | Typical Organisms / Infections | Examples |
|---|---|---|
| Antibody (B-cell / humoral) | Encapsulated bacteria (S. pneumoniae, H. influenzae), Giardia lamblia, enteroviruses | CVID, XLA, SAD |
| T-cell (cellular) | Viruses (CMV, EBV, VZV, adenovirus), fungi (Candida, PJP), intracellular bacteria (mycobacteria, Listeria) | SCID, DiGeorge, HIV |
| Phagocytes | Catalase-positive bacteria (S. aureus, Serratia, Burkholderia), Aspergillus, Nocardia | CGD, neutropenia |
| Complement (terminal) | Neisseria meningitidis, N. gonorrhoeae | C5–C9 deficiency |
| Complement (early/classical) | Encapsulated bacteria; immune-complex disease (SLE) | C1q, C2, C4 deficiency |
| Innate / interferon pathway | Mycobacteria (BCG disease), herpes viruses, intracellular bacteria | STAT1 GOF, IFN-γR deficiency |
Investigations
Investigation of suspected immunodeficiency should follow a stepwise approach — from basic screening tests available in primary care through to advanced functional and genetic assays performed in specialist centres. The Australasian Society of Clinical Immunology and Allergy (ASCIA) provides an investigation algorithm endorsed for Australian practice.
Tier 1 — Primary Care Screening (Available via MBS)
Tier 2 — Immunology Specialist Investigations
Tier 3 — Advanced / Genetic Testing
Management
Management of immunodeficiency is guided by the underlying mechanism, severity, and reversibility. The core principles are: (1) prevent and treat infections, (2) replace deficient immune components, (3) correct the underlying defect where possible, and (4) manage complications.
Immunoglobulin Replacement Therapy
Immunoglobulin replacement is the cornerstone of treatment for patients with primary antibody deficiency and selected patients with secondary hypogammaglobulinaemia (e.g., post-rituximab, CLL, myeloma).
Prophylactic Antimicrobials
Definitive Therapies for Severe Primary Immunodeficiency
| Therapy | Indication | Availability in Australia |
|---|---|---|
| Haematopoietic stem-cell transplant (HSCT) | SCID, CGD, Wiskott–Aldrich, HLH, severe CID — curative intent | Major paediatric transplant centres (RCH Melbourne, SCH Sydney, QCH Brisbane, PMH Perth). Matched sibling preferred; haploidentical increasingly available. |
| Gene therapy | ADA-SCID, X-linked SCID, WAS, CGD — emerging standard for selected PIDs | Clinical trials and compassionate access through specialised centres; not yet standard-of-care in Australia for most conditions. |
| Thymus transplantation | Complete DiGeorge syndrome (athymia) | Limited international centres; not currently performed in Australia — patients referred overseas. |
Vaccination Considerations
- Inactivated vaccines are generally safe and recommended, though efficacy may be reduced in immunodeficient patients.
- Household contacts of PID patients should be fully vaccinated, including live vaccines (with the exception of oral polio, which should be avoided in contacts of SCID patients).
- Influenza vaccine (inactivated) — recommended annually for all immunodeficient patients and their household contacts.
- COVID-19 vaccination — mRNA and protein subunit vaccines are safe; additional booster doses are recommended per ATAGI guidance for immunocompromised individuals.
- Pneumococcal vaccination — 23vPPV + PCV13 schedule recommended for antibody-deficient patients, though response may be suboptimal (document response).
- Post-immunoglobulin: Live vaccines should be deferred ≥8 months after IVIG/SCIG due to passive antibody interference; inactivated vaccines can be given but response assessment may be confounded.
Management of Specific Complications
| Complication | Association | Management |
|---|---|---|
| Bronchiectasis | CVID, XLA, other antibody deficiencies | Optimise immunoglobulin; chest physiotherapy; azithromycin maintenance; annual influenza + pneumococcal vaccination |
| Autoimmune cytopenias | CVID (30% of patients), ALPS | First-line: glucocorticoids; refractory: rituximab (with immunoglobulin monitoring), splenectomy (last resort) |
| GLILD (granulomatous-lymphocytic interstitial lung disease) | CVID | Azathioprine or mycophenolate +/− rituximab; multidisciplinary team management |
| Lymphoma | CVID, ALPS, Wiskott–Aldrich | Standard lymphoma protocols with immunology oversight; immunoglobulin replacement throughout treatment |
| Splenomegaly / granulomas | CVID, CGD | Avoid splenectomy if possible; immunomodulatory therapy for granulomas (steroids, TNF inhibitors with caution) |
Special Populations
Aboriginal and Torres Strait Islander peoples experience a disproportionate burden of infectious disease and secondary immunodeficiency-related complications. Structural inequities in healthcare access, higher prevalence of chronic conditions requiring immunosuppressive therapy, and the legacy of rheumatic heart disease (RHD) management all intersect with immunodeficiency care.
📚 References
- 1. Tangye SG, Al-Herz W, Bousfiha A, et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022;42(7):1473–1507.
- 2. Australasian Society of Clinical Immunology and Allergy (ASCIA). Primary Immunodeficiency (PID) Guide for Health Professionals. ASCIA; 2024. Available at: https://www.allergy.org.au.
- 3. Boyle JM, Buckley RH. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J Clin Immunol. 2007;27(5):497–502.
- 4. National Blood Authority Australia. Criteria for the Clinical Use of Intravenous Immunoglobulin in Australia. 3rd ed. NBA; 2012 (updated 2023).
- 5. Risma KA, Bhatt RR, Bhatt S, et al. COVID-19 outcomes in patients with primary immunodeficiency. J Allergy Clin Immunol Pract. 2022;10(3):762–771.
- 6. Patel SY, Carbone J, Jolles S. The expanding field of secondary antibody deficiency: causes, diagnosis, and management. Front Immunol. 2019;10:33.
- 7. Australian Technical Advisory Group on Immunisation (ATAGI). Australian Immunisation Handbook. Australian Government Department of Health; 2024. Available at: https://immunisationhandbook.health.gov.au.
- 8. AIHW (Australian Institute of Health and Welfare). Aboriginal and Torres Strait Islander Health Performance Framework: Immunisation. AIHW; 2023.
- 9. RHDAustralia (Rheumatic Heart Disease Australia). The 2020 Australian Guideline for Prevention, Diagnosis and Management of Acute Rheumatic Fever and Rheumatic Heart Disease. 3rd ed. Menzies School of Health Research; 2020.
- 10. Bousfiha A, Moundir A, Tangye SG, et al. The 2022 Update of IUIS Phenotypical Classification for Human Inborn Errors of Immunity. J Clin Immunol. 2022;42(5):1008–1020.
- 11. Bonilla FA, Khan DA, Ballas ZK, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol. 2015;136(5):1186–1205.
- 12. Phospholipidosis and related considerations. In: Australian Medicines Handbook (AMH). Adelaide: AMH Pty Ltd; 2024.