📋 Key Information Summary
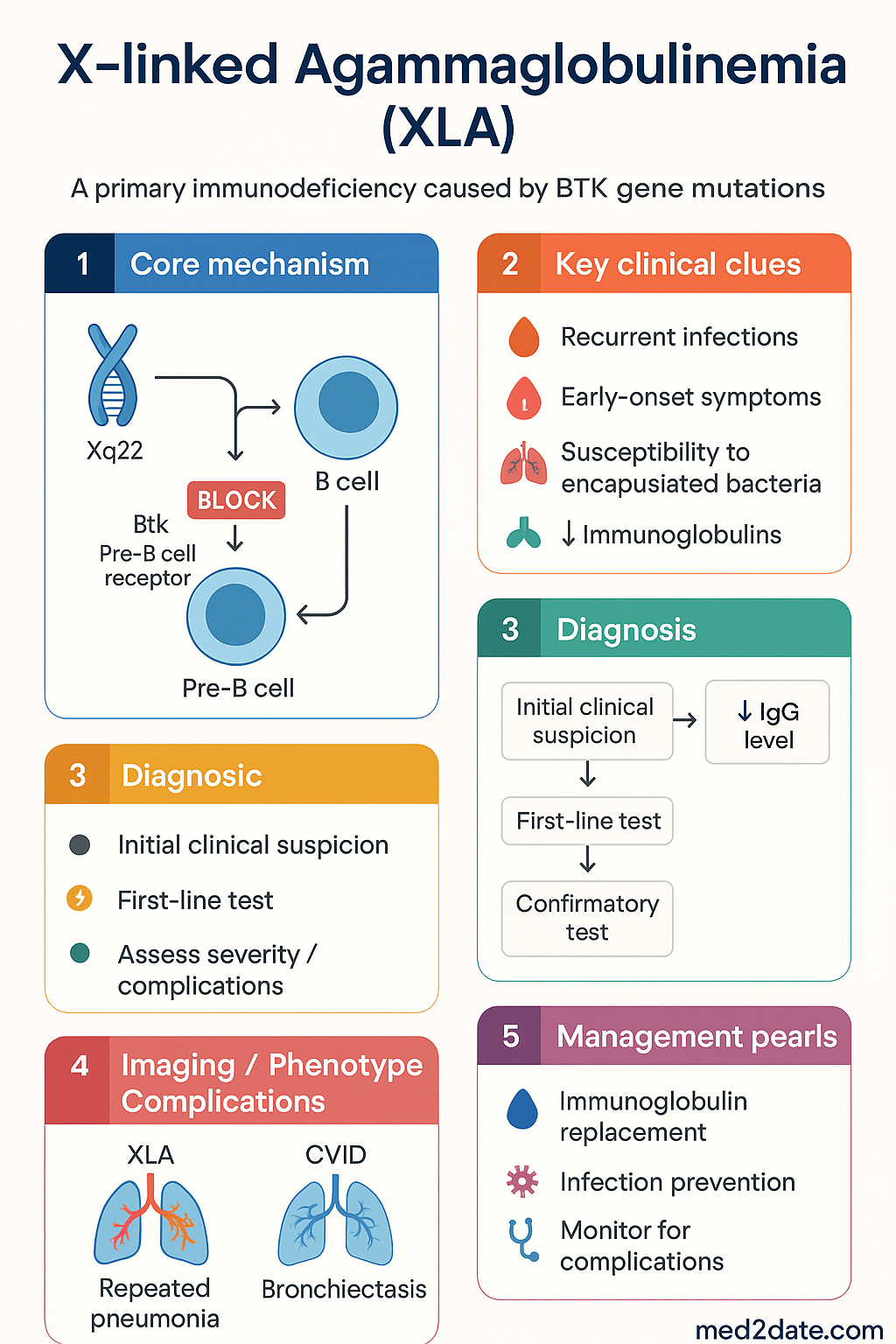
- X-linked agammaglobulinaemia (XLA) is a primary immunodeficiency caused by pathogenic variants in the BTK (Bruton tyrosine kinase) gene on Xq22, leading to a block in B-cell maturation at the pre-B stage.
- Affected males have a near-complete absence of mature B lymphocytes (<1 % of peripheral lymphocytes; absolute B-cell count <0.02 × 10⁹/L) and profoundly reduced serum immunoglobulins of all isotypes.
- Onset of recurrent sinopulmonary infections typically occurs after 6 months of age once passively transferred maternal IgG has waned.
- Encapsulated bacteria (Streptococcus pneumoniae, Haemophilus influenzae type b, Staphylococcus aureus) are the most common pathogens; enteroviral and Giardia infections also raise clinical suspicion.
- Diagnosis requires flow-cytometric demonstration of absent/reduced CD19⁺ B cells, profoundly low IgG (<2 g/L), IgA and IgM, plus confirmation by BTK gene sequencing.
- Immunoglobulin replacement therapy (IRT) — either intravenous (IVIG) every 3–4 weeks or subcutaneous (SCIG) weekly — is the cornerstone of lifelong management and is PBS Authority Required.
- Target trough IgG ≥ 8–10 g/L is associated with reduced pulmonary infections; some centres aim ≥ 10 g/L in patients with bronchiectasis.
- Live vaccines (OPV, BCG, MMR, varicella, yellow fever) are contraindicated; household contacts should not receive oral polio vaccine.
- Annual pulmonary function testing and HRCT are recommended to monitor for bronchiectasis, the major long-term complication.
- Haematopoietic stem cell transplantation (HSCT) is curative but generally reserved for patients with severe complications or poor IRT response; it is performed at major Australian paediatric centres (Melbourne, Sydney).
- Carrier females are usually asymptomatic but may have skewed X-inactivation causing mild hypogammaglobulinaemia in approximately 10–15 %.
- Genetic counselling should be offered to all families; prenatal and preimplantation genetic diagnosis are available in Australian centres.
- ATSI communities face additional barriers to timely diagnosis and IRT access; engagement with Indigenous liaison officers and RFDS-supported SCIG programmes is essential.
- Prognosis with early diagnosis, adequate IRT, and proactive lung surveillance has improved substantially; many patients now survive into adulthood with preserved lung function.
Introduction & Australian Epidemiology
X-linked agammaglobulinaemia (XLA; OMIM #300755), first described by Colonel Ogden Bruton in 1952, is the prototypical primary antibody deficiency. It accounts for approximately 85 % of all cases of agammaglobulinaemia and represents roughly 6–9 % of all diagnosed primary immunodeficiencies (PIDs) worldwide.
The disorder results from pathogenic variants in the BTK (Bruton tyrosine kinase) gene located on chromosome Xq22.1. Over 1 600 unique mutations have been catalogued (BTKbase, last updated 2024), with no single hotspot mutation predominating, although missense mutations in the kinase domain are the most frequently identified class.
Australian incidence: Based on data from the Australian Primary Immunodeficiency (AusPID) registry and the Australasian Society of Clinical Immunology and Allergy (ASCIA), the estimated birth prevalence of XLA in Australia is approximately 1 in 100 000–200 000 live male births. In practice, this translates to approximately 1–3 new diagnoses per year nationally. Median age at diagnosis remains 2–3 years, although early genomic testing is reducing this interval in families with known variants.
XLA affects all ethnic groups and geographic regions. There is no convincing evidence of founder effects in Australian populations, although consanguinity in some communities may elevate carrier frequency. Males of any ancestry are equally at risk.
BTK Mutation & B-Cell Development
The BTK Gene and Protein
Bruton tyrosine kinase (BTK) is a 659-amino-acid non-receptor tyrosine kinase belonging to the Tec kinase family. The gene spans 37.5 kb of genomic DNA comprising 19 exons. BTK contains five functional domains:
- PH (pleckstrin homology) domain — binds phosphatidylinositol-3,4,5-trisphosphate (PIP₃), recruiting BTK to the plasma membrane.
- TH (Tec homology) domain — contains a zinc-finger motif involved in protein–protein interactions.
- SH3 domain — mediates intramolecular and intermolecular interactions.
- SH2 domain — recognises phosphotyrosine motifs on adaptor proteins.
- Kinase (SH1) domain — catalytic domain responsible for phosphorylation of downstream substrates (PLCγ2, NF-κB pathway components).
Mutation Spectrum
| Mutation Type | Approximate Frequency | Typical Phenotype |
|---|---|---|
| Missense | ~40 % | Variable — some residual BTK activity; milder clinical course possible |
| Nonsense / Frameshift | ~35 % | Typically classic severe XLA; absent or truncated protein |
| Splice-site | ~15 % | Variable; depends on exon skipped |
| Large deletions / Intron mutations | ~10 % | Usually severe; may escape detection by conventional Sanger sequencing |
Normal B-Cell Development & the BTK Block
In healthy individuals, B-cell lymphopoiesis proceeds through ordered stages in the bone marrow:
- Pro-B cell → D-J then V-DJ rearrangement of the immunoglobulin heavy chain (IgH).
- Pre-B cell — expresses μ heavy chain with surrogate light chain (VpreB/λ5) as the pre-B-cell receptor (pre-BCR). This is the stage where BTK signalling is essential.
- Immature B cell — expresses surface IgM after successful light-chain rearrangement.
- Mature naïve B cell — co-expresses IgM and IgD; emigrates to secondary lymphoid organs.
BTK is activated downstream of the pre-BCR and BCR via Lyn/Syk kinases → BTK autophosphorylation (Y551) → PLCγ2 activation → calcium flux and NF-κB signalling. Without functional BTK, pre-B cells fail to receive survival and proliferative signals, leading to developmental arrest at the pre-B stage and subsequent apoptosis. Consequently, affected males have <1 % mature B lymphocytes in peripheral blood.
T-cell immunity is preserved because BTK is not expressed in the T-cell lineage. Therefore, delayed-type hypersensitivity responses, viral clearance (except enteroviruses), and tumour immunosurveillance by T cells remain largely intact.
Clinical Presentation
Typical Presentation Timeline
Infection Spectrum
| Site | Common Pathogens | Notes |
|---|---|---|
| Upper respiratory | S. pneumoniae, H. influenzae, M. catarrhalis | Otitis media, sinusitis — most common initial presentation |
| Lower respiratory | S. pneumoniae, H. influenzae, S. aureus, Pseudomonas, Mycoplasma | Pneumonia → bronchiectasis if undertreated |
| Gastrointestinal | Giardia lamblia, enteroviruses, Campylobacter | Chronic diarrhoea, malabsorption |
| CNS | Enteroviruses (esp. ECHO), Poliovaccine strains | Enteroviral meningoencephalitis — high mortality if acquired |
| Skin / Soft tissue | S. aureus, dermatophytes | Pyoderma, abscess formation |
| Musculoskeletal | Mycoplasma, Ureaplasma | Septic arthritis — unusual in immunocompetent hosts |
| Haematological | Parvovirus B19 | Transient aplastic crisis; chronic pure red cell aplasia reported |
Non-Infectious Complications
- Bronchiectasis — the most significant long-term complication, occurring in 30–50 % of patients who experienced a delay in diagnosis. Irreversible once established.
- Autoimmune disease — reported in 10–20 % of XLA patients, including inflammatory arthritis, autoimmune haemolytic anaemia, and inflammatory bowel disease.
- Malignancy — increased risk of lymphoma (particularly Burkitt and DLBCL subtypes) and colorectal cancer in adult XLA patients.
- Growth retardation — secondary to chronic infection and malabsorption.
- Enteroviral dermatomyositis-like syndrome — a distinctive, often fatal manifestation of chronic enteroviral infection in untreated XLA patients.
Diagnosis
Diagnostic Algorithm
Investigations Summary
Diagnostic Criteria (IUIS 2022)
- Definite XLA: Male with absent/reduced CD19⁺ B cells (< 2 %), profoundly low IgG + IgA + IgM, AND a pathogenic or likely pathogenic BTK variant identified.
- Probable XLA: Male with absent B cells + profoundly low immunoglobulins + absent/abnormal BTK protein expression, WITHOUT a confirmed mutation (possible deep intronic or regulatory variant).
- Carrier detection: Females with a family history may show non-random X-inactivation patterns; however, carrier testing by X-chromosome inactivation assay is not routinely recommended — direct mutation detection is preferred.
IVIG Treatment & Prognosis
Immunoglobulin Replacement Therapy (IRT)
Lifelong immunoglobulin replacement is the standard of care for XLA and is PBS Authority Required. IRT provides passive humoral immunity, substantially reducing the frequency and severity of bacterial infections.
IgG Trough Level Targets
| IgG Trough Target | Clinical Context |
|---|---|
| ≥ 5 g/L | Absolute minimum; below this level, infection risk rises sharply |
| ≥ 8 g/L | Standard target for most patients; associated with significant reduction in pneumonia |
| ≥ 10 g/L | Preferred target in patients with established bronchiectasis, structural lung disease, or refractory infections |
Monitoring on IRT
- Check IgG trough level before each infusion (for IVIG) or quarterly (for SCIG). Aim ≥ 8 g/L.
- Annual spirometry (from age 6) and HRCT every 2–5 years or if FEV₁ declines.
- Annual complete blood count — monitor for parvovirus-related anaemia, lymphoproliferative disease.
- Liver function tests and hepatitis C PCR (historically relevant; modern IG products have negligible risk).
- Document infection frequency: ≥ 3 significant bacterial infections/year despite trough ≥ 8 g/L warrants dose escalation or evaluation for complications.
Adverse Effects of IVIG
- Common (1–15 %): Headache, chills, myalgia, flushing, back pain — usually managed by slowing infusion rate and pre-medication with paracetamol ± antihistamine.
- Rare (<1 %): Anaphylaxis (especially in IgA-deficient patients receiving IgA-containing products), aseptic meningitis, thromboembolic events, haemolytic anaemia (anti-A/anti-B antibodies in some products), acute kidney injury (sucrose-containing products).
Antibiotic Prophylaxis & Acute Infection Management
- Long-term prophylaxis: Many immunologists recommend low-dose oral antibiotics (e.g., amoxicillin 250–500 mg PO daily in adults; 15–25 mg/kg/day in children, PBS General Benefit) or azithromycin (500 mg PO three times per week) for patients with recurrent sinopulmonary infections or bronchiectasis, per eTG Antibiotic guidelines.
- Acute infections: Treat promptly and aggressively with appropriate antibiotics. Lower threshold for IV antibiotics and hospital admission compared to immunocompetent patients. Consider broader coverage (including atypical organisms) in respiratory infections.
- Giardia: Metronidazole 400 mg PO TDS for 5–7 days (adults) or tinidazole 50 mg/kg (max 2 g) as a single dose. PBS General Benefit.
Curative Therapy — Haematopoietic Stem Cell Transplantation (HSCT)
HSCT can reconstitute the B-cell lineage and is potentially curative. In Australia, it is considered when:
- Progressive bronchiectasis despite optimised IRT (trough IgG consistently ≥ 10 g/L).
- Severe enteroviral infections unresponsive to IVIG and antiviral therapy.
- Significant autoimmune complications.
- A matched sibling donor (MSD) is available — outcomes with MSD HSCT are excellent (>90 % survival).
Australian centres performing HSCT for PID include the Royal Children's Hospital Melbourne, The Children's Hospital at Westmead (Sydney), and Queensland Children's Hospital (Brisbane). Referral to a paediatric immunology/transplant team is mandatory.
Emerging Therapies
- Gene therapy: Preclinical and early-phase clinical trials are investigating lentiviral vector-mediated BTK gene transfer into haematopoietic stem cells. Not yet available in Australia outside clinical trials.
- Small-molecule BTK modulators: Primarily studied in B-cell malignancies; potential future role in modulating residual BTK activity in hypomorphic mutations.
Prognosis
With early diagnosis and adequate immunoglobulin replacement, the prognosis for XLA has improved dramatically over the past three decades. Australian registry data indicate:
- Patients diagnosed before 2 years of age and commenced on IRT promptly have near-normal life expectancy.
- The strongest predictor of long-term lung health is the cumulative burden of lower respiratory tract infections before IRT commencement — reinforcing the importance of early diagnosis.
- Patients diagnosed late (after age 5) or with delayed IRT initiation have significantly higher rates of bronchiectasis and reduced lung function.
- Survival into the 6th and 7th decades of life has been documented in well-managed patients.
- Quality of life is generally good, particularly with SCIG home therapy. Psychological support should be offered to adolescents transitioning to independent care.
Special Populations
ATSI Health Considerations
While XLA has no known increased prevalence in Aboriginal and Torres Strait Islander populations, the higher background burden of respiratory infections in ATSI children creates diagnostic and management challenges that require culturally safe, community-centred approaches.
📚 References
- 1. Bruton OC. Agammaglobulinemia. Pediatrics. 1952;9(6):722–728.
- 2. Conley ME, Broides A, Hernandez-Trujillo V, et al. Genetic analysis of patients with defects in early B-cell development. Immunol Rev. 2005;203:216–234.
- 3. Tangye SG, Al-Herz W, Bousfiha A, et al. Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022;42(7):1473–1507.
- 4. Bousfiha A, Jeddane L, Picard C, et al. The 2017 IUIS phenotypical classification for primary immunodeficiencies. J Clin Immunol. 2018;38(1):129–143.
- 5. Winkelstein JA, Marino MC, Lederman HM, et al. X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine (Baltimore). 2006;85(4):193–202.
- 6. El-Sayed ZA, Abramova I, Aldave JC, et al. X-linked agammaglobulinemia (XLA): phenotype, diagnosis, and therapeutic challenges around the world. World Allergy Organ J. 2019;12(3):100018.
- 7. Australasian Society of Clinical Immunology and Allergy (ASCIA). ASCIA Primary Immunodeficiency (PID) Guide for Health Professionals. Sydney: ASCIA; 2023. Available at: www.allergy.org.au.
- 8. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander Health Performance Framework 2020: Summary report. Canberra: AIHW; 2020.
- 9. Chapel H, Lucas M, Lee M, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112(2):277–286.
- 10. Hernandez-Trujillo VP, Scalchunes C, Cunningham-Rundles C, et al. Autoimmunity and inflammation in X-linked agammaglobulinemia. J Clin Immunol. 2014;34(6):627–632.
- 11. Quinti I, Soresina A, Agostini C, et al. Prospective study on CVID patients with adverse reactions to intravenous or subcutaneous immunoglobulin replacement. Allergy. 2018;73(1):201–208.
- 12. Gennery AR, Slatter MA, Bhatt J, et al. Hematopoietic stem cell transplantation in patients with primary antibody deficiency. J Allergy Clin Immunol. 2020;145(5):1453–1462.
- 13. National Health and Medical Research Council (NHMRC). Australian Immunisation Handbook. Canberra: Australian Government Department of Health; 2024. Available at: immunisationhandbook.health.gov.au.
- 14. Väliaho J, Faisal I, Ortutay C, et al. Mutation spectrum and phenotype–genotype correlation in X-linked agammaglobulinemia (BTKbase update). Hum Mutat. 2020;41(8):1402–1416.
- 15. RHDAustralia. Australian Guidelines for the Prevention and Control of Infection in Healthcare. Canberra: NHMRC; 2019 (updated 2024).