📋 Key Information Summary
- Primary immunodeficiencies (PIDs) are a heterogeneous group of >480 monogenic disorders affecting innate or adaptive immunity, with an estimated prevalence of 1 in 1,200–2,000 Australians.
- PIDs follow autosomal dominant, autosomal recessive, X-linked, or polygenic inheritance; X-linked conditions disproportionately affect males.
- The "10 Warning Signs of PID" (Jeffrey Modell Foundation) should be applied in any child or adult with recurrent, severe, or unusual infections.
- Combined immunodeficiencies (e.g., Severe Combined Immunodeficiency — SCID) are medical emergencies; newborn screening by TREC assay is now routine in Australia.
- Antibody deficiencies (CVID, XLA) are the most common symptomatic PIDs and present with recurrent sinopulmonary infections from late childhood or early adulthood.
- Diagnosis requires immunoglobulin quantification, lymphocyte subset analysis, functional immune assays, and increasingly next-generation sequencing (genomic testing).
- Immunoglobulin replacement therapy (IVIg or SCIg) is the mainstay for antibody-deficient patients and is PBS-authority listed in Australia.
- Haematopoietic stem cell transplantation (HSCT) is curative for SCID and other severe PIDs and is available at specialised paediatric centres in Sydney, Melbourne, Brisbane, and Perth.
- Gene therapy is emerging for ADA-SCID, X-linked SCID, and Wiskott–Aldrich syndrome; clinical trials are available in Australia.
- Genetic counselling is essential for all families with confirmed PIDs; pre-symptomatic diagnosis in subsequent pregnancies can be achieved by cord-blood or prenatal genetic testing.
- Live vaccines (BCG, OPV, MMR, varicella, yellow fever) are contraindicated in suspected or confirmed severe PIDs until immunological status is clarified.
- Aboriginal and Torres Strait Islander communities face higher infectious burdens and reduced access to specialist immunology services, necessitating outreach and telehealth pathways.
Introduction & Australian Epidemiology
Primary immunodeficiencies (PIDs) are inborn errors of immunity caused by single-gene (monogenic) or, less commonly, polygenic mutations that compromise one or more arms of the immune system. The International Union of Immunological Societies (IUIS) 2022 classification recognises over 480 distinct disorders, categorised by the affected immune compartment: combined T- and B-cell defects, predominantly antibody deficiencies, phagocyte defects, complement deficiencies, immune dysregulation syndromes, autoinflammatory disorders, and innate immune defects.
In Australia, the Australasian Society of Clinical Immunology and Allergy (ASCIA) and the Australian Institute of Health and Welfare (AIHW) estimate that approximately 25,000–50,000 Australians live with a PID, though many remain undiagnosed. The Australian National PID Registry (managed through ASCIA) has enrolled several thousand patients since its inception. Antibody deficiencies account for approximately 55–65 % of diagnosed PIDs, followed by phagocytic disorders (~15 %), combined immunodeficiencies (~10 %), complement deficiencies (~5 %), and other disorders.
The advent of newborn screening using the T-cell receptor excision circle (TREC) assay, implemented nationally in Australia from 2018–2020, has transformed early detection of SCID and other severe T-cell lymphopenias. This has significantly reduced mortality by enabling timely referral for definitive therapy such as HSCT.
Early recognition and appropriate management of PIDs remain critical, as delayed diagnosis is associated with irreversible organ damage, chronic lung disease (bronchiectasis), and increased mortality. This guideline outlines the genetic mechanisms, classification, diagnostic approach, and treatment strategies for inherited immune deficiencies in the Australian context.
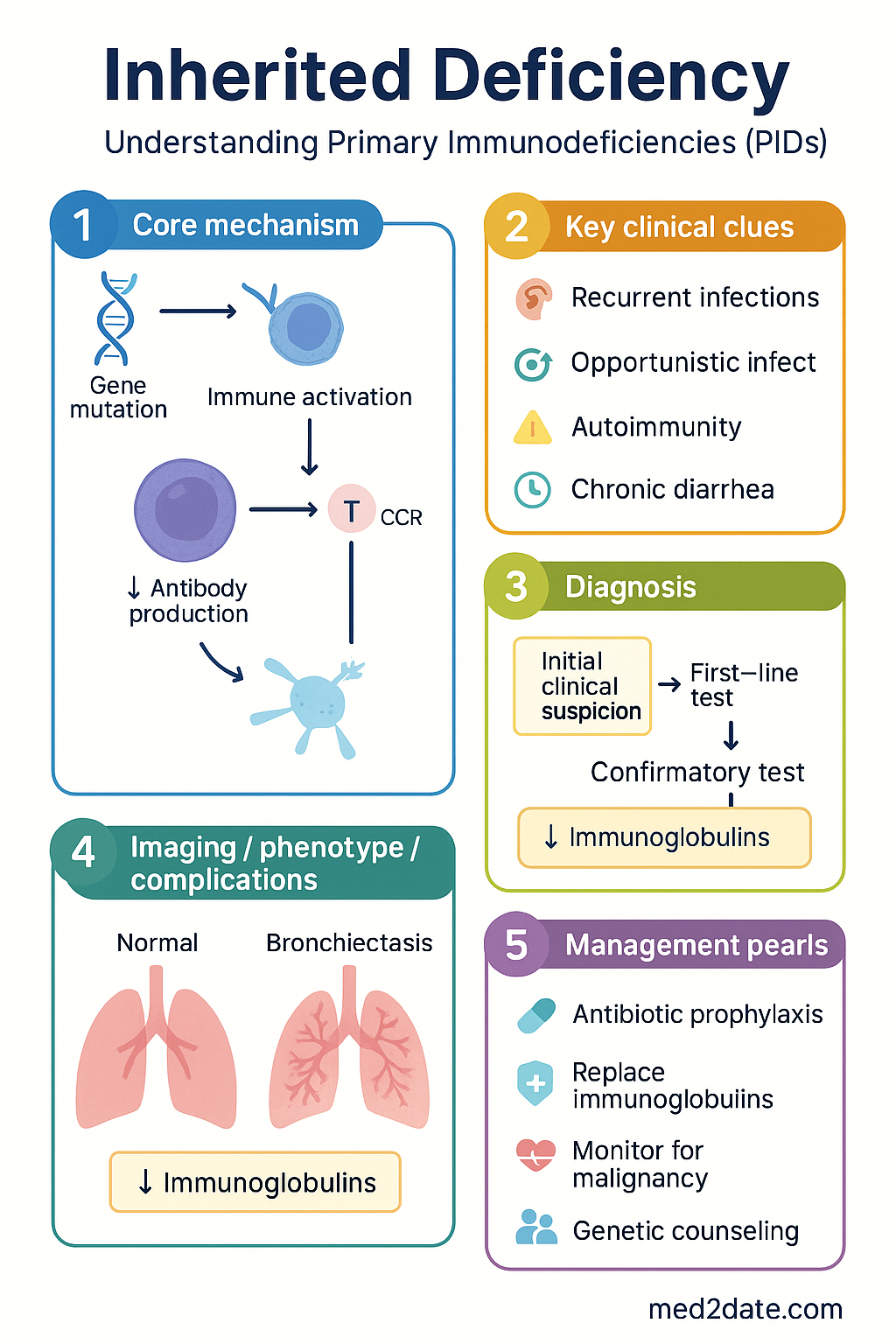
Genetic Mechanisms
Understanding the genetic basis of PIDs is essential for accurate diagnosis, prognostication, and family counselling. The following genetic mechanisms underpin the majority of inherited immune deficiencies.
Inheritance Patterns
| Pattern | Examples | Key Features |
|---|---|---|
| X-linked recessive | XLA (BTK), X-SCID (IL2RG), Wiskott–Aldrich (WAS), CGD (CYBB) | Males predominantly affected; carrier females usually asymptomatic; 50 % of sons of carrier mothers affected |
| Autosomal recessive | ADA-SCID, PNP deficiency, Artemis-SCID, RAG1/2 deficiency, MHC II deficiency | Both parents carriers; 25 % risk per pregnancy; consanguinity increases risk significantly |
| Autosomal dominant | STAT1 GOF, STAT3 LOF (Hyper-IgE syndrome), CTLA-4 haploinsufficiency | 50 % transmission; variable penetrance and expressivity |
| Somatic (de novo) | Activated PI3Kδ syndrome (somatic PIK3CD mutations) | Not inherited; present in only a proportion of lymphocytes |
| Polygenic / complex | Selective IgA deficiency, partial complement deficiencies | Multiple loci contribute; variable expressivity; may progress to CVID |
Molecular Pathways Affected
- T-cell development: Mutations in IL2RG, JAK3, RAG1/2, ADA, DCLRE1C (Artemis), and IL7RA disrupt thymic T-cell maturation, resulting in severe combined immunodeficiency.
- B-cell differentiation: BTK mutations prevent pre-B-cell receptor signalling (XLA); defects in ICOS, TACI, BAFF-R, or CD19 impair class-switch recombination and somatic hypermutation (CVID phenocopies).
- Phagocyte function: NADPH oxidase complex mutations (CYBB, CYBA, NCF1, NCF2, NCF4) cause chronic granulomatous disease (CGD); mutations in ELANE or HAX1 cause severe congenital neutropaenia.
- Complement cascade: Deficiencies in early classical pathway components (C1q, C2, C4) predispose to immune-complex disease; terminal complement defects (C5–C9) predispose to Neisseria meningitidis infections.
- Innate signalling: Defects in TLR3 (herpes simplex encephalitis), IFN-γ/IL-12 axis (Mendelian susceptibility to mycobacterial disease), and complement lectin pathway.
Types of Deficiency
PIDs are classified according to the IUIS 2022 framework. The following table summarises the major categories with representative Australian-relevant examples.
Combined Immunodeficiencies
| Disorder | Gene(s) | Inheritance | Key Features |
|---|---|---|---|
| X-linked SCID | IL2RG | X-linked | Absent T cells, present B cells (T⁻B⁺NK⁻); most common SCID subtype |
| ADA-SCID | ADA | AR | T⁻B⁻NK⁻; toxic metabolite accumulation; skeletal abnormalities |
| Artemis-SCID | DCLRE1C | AR | Radiosensitive; T⁻B⁻NK⁺; higher prevalence in some First Nations populations |
| Omenn syndrome | RAG1/2, DCLRE1C | AR | Erythroderma, lymphadenopathy, hepatosplenomegaly, elevated IgE |
| MHC II deficiency | CIITA, RFXANK, RFX5, RFXAP | AR | CD4⁺ T-cell lymphopenia; severe diarrhoea and respiratory infections |
Predominantly Antibody Deficiencies
| Disorder | Gene(s) | Inheritance | Key Features |
|---|---|---|---|
| X-linked agammaglobulinaemia (XLA) | BTK | X-linked | Absent B cells; markedly reduced all immunoglobulin classes; presents 6–12 months |
| Common variable immunodeficiency (CVID) | Often polygenic; ICOS, TACI, BAFF-R in some | Variable | Most common symptomatic PID in adults; reduced IgG ± IgA/IgM; autoimmunity, granulomata |
| Selective IgA deficiency | Largely unknown | Polygenic | IgA <0.07 g/L; most common PID overall (~1:300–700); often asymptomatic |
| Hyper-IgM syndromes | CD40LG, CD40, AID, UNG | X-linked (CD40LG) or AR | Defective class-switch recombination; normal/elevated IgM, low IgG/IgA/IgE; opportunistic infections |
Phagocytic Disorders
- Chronic granulomatous disease (CGD): Defective NADPH oxidase → inability to kill catalase-positive organisms (Staphylococcus aureus, Aspergillus, Serratia, Burkholderia). X-linked (CYBB) ~70 %; autosomal recessive ~30 %. Characterised by granuloma formation, lymphadenitis, liver abscess, and pulmonary aspergillosis.
- Severe congenital neutropaenia (SCN): ELANE, HAX1, and other gene mutations. ANC persistently <0.2 × 10⁹/L. High risk of bacterial sepsis and myelodysplasia/leukaemia if untreated.
- Leukocyte adhesion deficiency (LAD) I–III: Impaired neutrophil migration. LAD-I (ITGB2/CD18) most common — delayed umbilical cord separation, poor wound healing, absent pus formation.
Complement Deficiencies
- Early classical (C1q, C2, C4): Strongly associated with SLE-like illness and immune-complex glomerulonephritis.
- Terminal complement (C5–C9): Recurrent Neisseria meningitidis infections; meningococcal vaccination essential.
- Mannose-binding lectin (MBL) deficiency: Common polymorphism (~5 % homozygous); clinical significance debated but may increase infection susceptibility in early childhood.
Immune Dysregulation Syndromes
- IPEX (FOXP3): X-linked; severe autoimmune enteropathy, eczema, type 1 diabetes, thyroiditis in infancy.
- IPEX-like (CTLA-4, LRBA, STAT1 GOF): Autoimmune cytopenias, lymphoproliferation, organ-specific autoimmunity.
- ALPS (FAS, FASLG, CASP10): Defective lymphocyte apoptosis → massive lymphadenopathy, splenomegaly, autoimmune cytopenias, elevated DNT cells.
Autoinflammatory Disorders
- Cryopyrin-associated periodic syndromes (CAPS): NLRP3 mutations; urticaria-like rash, fever, arthralgia, meningitis. Responsive to IL-1 blockade (anakinra, canakinumab).
- Familial Mediterranean fever (FMF): MEFV mutations; recurrent febrile serositis; colchicine prophylaxis.
- TRAPS: TNFRSF1A mutations; prolonged febrile episodes with periorbital oedema and myalgia.
Diagnosis
When to Suspect a PID
The Jeffrey Modell Foundation "10 Warning Signs" remain a useful screening tool. Any patient meeting ≥2 criteria warrants immunological evaluation:
- ≥4 new ear infections within 1 year
- ≥2 serious sinus infections within 1 year
- ≥2 months on antibiotics with little effect
- ≥2 pneumonias within 1 year
- Failure to thrive or poor growth
- Recurrent deep-seated infections (liver/brain abscess, osteomyelitis)
- Persistent thrush or fungal skin infection beyond infancy
- Need for IV antibiotics to clear infections
- ≥2 family members with PID
- Features suggestive of immune dysregulation (autoimmune cytopenias, granulomata, severe eczema)
First-Line Investigations (Primary Care)
Second-Line Investigations (Specialist Immunology)
Newborn Screening — TREC Assay
Diagnostic Algorithm
Genetic Counselling & Treatment
Genetic Counselling
All families with a confirmed molecular diagnosis of PID should be referred to a clinical geneticist or genetic counsellor. Key counselling components include:
- Recurrence risk education: X-linked — 50 % carrier risk for daughters, 50 % affected sons; autosomal recessive — 25 % per pregnancy; autosomal dominant — 50 % with variable penetrance.
- Carrier testing: Available for X-linked conditions (BTK, IL2RG, WAS) and most autosomal recessive PIDs. Pre-conception carrier screening programmes (e.g., Mackenzie's Mission) are expanding in Australia.
- Prenatal diagnosis: Chorionic villus sampling (CVS) at 11–13 weeks or amniocentesis at 15–18 weeks for known familial mutations. Non-invasive prenatal testing (NIPT) is not yet standard for PIDs.
- Preimplantation genetic testing (PGT-M): Available at accredited Australian IVF centres for families with known monogenic PID mutations.
- Psychosocial support: Referral to patient organisations (IDFA — Immune Deficiencies Foundation of Australia; Jeffrey Modell Foundation ANZ) for peer support and advocacy.
Treatment Overview
Immunoglobulin Replacement Therapy
The cornerstone of management for antibody-deficient patients. Two routes are available in Australia:
Antibiotic Prophylaxis
- Co-trimoxazole (trimethoprim–sulfamethoxazole) 480 mg PO daily (paediatric: 5–6 mg/kg/day trimethoprim component) — first-line for CGD prophylaxis and adjunctive in antibody deficiency. PBS: General Benefit.
- Azithromycin 250 mg PO three times weekly (paediatric: 5–10 mg/kg/week) — alternative or adjunct for CGD and bronchiectasis prophylaxis in CVID. PBS: Authority Required for prophylaxis indications.
- Amoxicillin 500 mg PO daily or cephalexin 500 mg PO daily — adjunctive in specific antibody deficiency with recurrent URTI. PBS: General Benefit.
Definitive Therapies
| Therapy | Indication | Availability in Australia |
|---|---|---|
| Haematopoietic stem cell transplant (HSCT) | SCID (all forms), WAS, CGD (severe), HLH, IPEX | Sydney Children's Hospital, Royal Children's Hospital Melbourne, Queensland Children's Hospital, Perth Children's Hospital |
| Gene therapy | ADA-SCID (Strimvelis® / OTL-101), X-SCID, WAS | Clinical trials at RCH Melbourne; some patients referred overseas |
| Enzyme replacement (PEG-ADA) | ADA-SCID (bridge to HSCT or if HSCT not feasible) | PBS Authority Required; administered IV twice weekly |
| Granulocyte colony-stimulating factor (G-CSF) | Severe congenital neutropaenia (ELANE, HAX1) | Filgrastim (Neupogen®) PBS General Benefit; pegfilgrastim available |
| Biologic therapies (IL-1, IL-6, JAK inhibitors) | Autoinflammatory disorders (CAPS, TRAPS), immune dysregulation | Anakinra (Kineret®) PBS Authority for CAPS; tocilizumab, baricitinib on case-by-case |
Infection Prevention & Vaccination
- All inactivated vaccines are safe and recommended (including COVID-19, influenza, pneumococcal conjugate, meningococcal B and ACWY).
- Live vaccines are CONTRAINDICATED in SCID, combined immunodeficiencies, symptomatic XLA/CVID, and phagocytic disorders until immunological clearance is obtained.
- BCG: Contraindicated in all severe PIDs. Neonates from high-risk TB settings should have immune evaluation before BCG administration.
- MMR and varicella vaccines: May be safe in mild/moderate PIDs (e.g., selective IgA deficiency, specific antibody deficiency) — specialist advice required.
- Household contacts of PID patients should be fully vaccinated, including annual influenza vaccination.
Monitoring
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Tangye SG, Al-Herz W, Bousfiha A, et al. Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022;42(7):1473–1507.
- 2. Australasian Society of Clinical Immunology and Allergy (ASCIA). Primary immunodeficiency (PID) guide for healthcare professionals. ASCIA; 2024. Available at: https://www.allergy.org.au
- 3. Boyle JM, Buckley RH. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J Clin Immunol. 2007;27(5):497–502.
- 4. Kwan A, Abraham RS, Currier R, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. 2014;312(7):729–738.
- 5. Australian Government Department of Health and Aged Care. Newborn bloodspot screening national policy framework. Commonwealth of Australia; 2023.
- 6. Immune Deficiencies Foundation of Australia (IDFA). Living with primary immunodeficiency in Australia. IDFA; 2023. Available at: https://idfa.org.au
- 7. Bonilla FA, Khan DA, Ballas ZK, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol. 2015;136(5):1186–1205.
- 8. Perez EE, Orange JS, Bonilla F, et al. Update on the use of immunoglobulin in human disease: a review of evidence. J Allergy Clin Immunol. 2017;139(3S):S1–S46.
- 9. Australian Institute of Health and Welfare (AIHW). Chronic respiratory conditions: bronchiectasis in Aboriginal and Torres Strait Islander people. AIHW; 2023. Cat. no. PHE 312.
- 10. Kirkpatrick P, Riminton S. Primary immunodeficiency diseases in Australia and New Zealand. J Clin Immunol. 2017;37(3):291–297.
- 11. National Health and Medical Research Council (NHMRC). Australian guidelines for the clinical care of people with cystic fibrosis 2023 (immunological considerations). NHMRC; 2023.
- 12. Mackenzie's Mission. Reproductive genetic carrier screening programme. Australian Genomics; 2023. Available at: https://www.mackenziesmission.org.au