📋 Key Information Summary
- Hypogammaglobulinemia denotes a serum immunoglobulin level >2 SD below the age-matched mean, resulting in impaired humoral immunity and recurrent infections — predominantly sinopulmonary, gastrointestinal, and skin/soft tissue.
- The most prevalent primary immunodeficiency worldwide and in Australia is Common Variable Immunodeficiency (CVID), with an estimated prevalence of 1 in 25,000.
- X-linked Agammaglobulinemia (XLA, Bruton disease) affects approximately 1 in 200,000 male births; absent mature B cells (CD19⁺) and profoundly low all immunoglobulin isotypes.
- Secondary causes — haematological malignancy, nephrotic syndrome, protein-losing enteropathy, immunosuppressive therapy — are more common in general practice than primary defects.
- Suspect hypogammaglobulinemia when a patient has ≥2 significant bacterial infections per year, ≥1 episode of pneumonia per year, or an opportunistic infection with encapsulated organisms.
- Diagnostic triad: total IgG <5 g/L, impaired specific antibody responses to protein (tetanus) and polysaccharide (pneumococcal) vaccines, and exclusion of secondary causes.
- Immunoglobulin replacement therapy (IgRT) — either intravenous (IVIG) every 3–4 weeks or subcutaneous (SCIG) weekly — is the cornerstone of treatment, aiming for a trough IgG ≥7 g/L.
- IVIG in Australia is available under PBS Authority Required listing; prescribers must apply through the PBS Immunoglobulin Governance Programme via the National Blood Authority (NBA).
- SCIG is increasingly preferred in Australia, enabling self-administration and reducing hospital attendance — particularly important for patients in regional and remote areas.
- Antibiotic prophylaxis (e.g. amoxicillin or azithromycin) is adjunctive, not a substitute for IgRT; breakthrough infections prompt re-evaluation of trough levels and adherence.
- Monitor trough IgG every 3–6 months initially, then annually; assess lung function (spirometry, DLCO) and high-resolution CT chest for bronchiectasis at baseline and every 1–2 years.
- Aboriginal and Torres Strait Islander peoples experience higher rates of chronic lung disease and reduced access to immunology services; early referral from rural and remote communities is critical.
- Live vaccines (MMR, varicella, oral polio, BCG) are contraindicated in patients with severe hypogammaglobulinemia; all household contacts should receive standard immunisations including influenza and pneumococcal vaccines.
Introduction & Australian Epidemiology
Hypogammaglobulinemia is characterised by low levels of one or more classes of serum immunoglobulins, leading to impaired humoral immunity and increased susceptibility to recurrent bacterial, viral, and fungal infections. It may arise as a primary (inherited) immunodeficiency or develop secondary to haematological, renal, gastrointestinal, or iatrogenic causes.
In Australia, the Australasian Society of Clinical Immunology and Allergy (ASCIA) estimates that primary immunodeficiencies affect approximately 1 in 1,200 to 1 in 2,000 individuals, though significant under-diagnosis persists. CVID is the most frequently diagnosed symptomatic primary antibody deficiency in adults, with onset typically in the second to fourth decade. XLA presents in infancy and early childhood, typically after the waning of maternal IgG at 6–9 months of age.
The AIHW National Hospital Morbidity Database shows that infections attributable to immune deficiency contribute to substantial hospitalisations, particularly in paediatric and elderly populations. Referral pathways through state-based immunology centres (e.g. Royal Adelaide Hospital Immunology, Westmead Hospital Immunology, Alfred Health Immunology, Royal Melbourne Hospital) facilitate access to specialist investigation and IgRT governance.
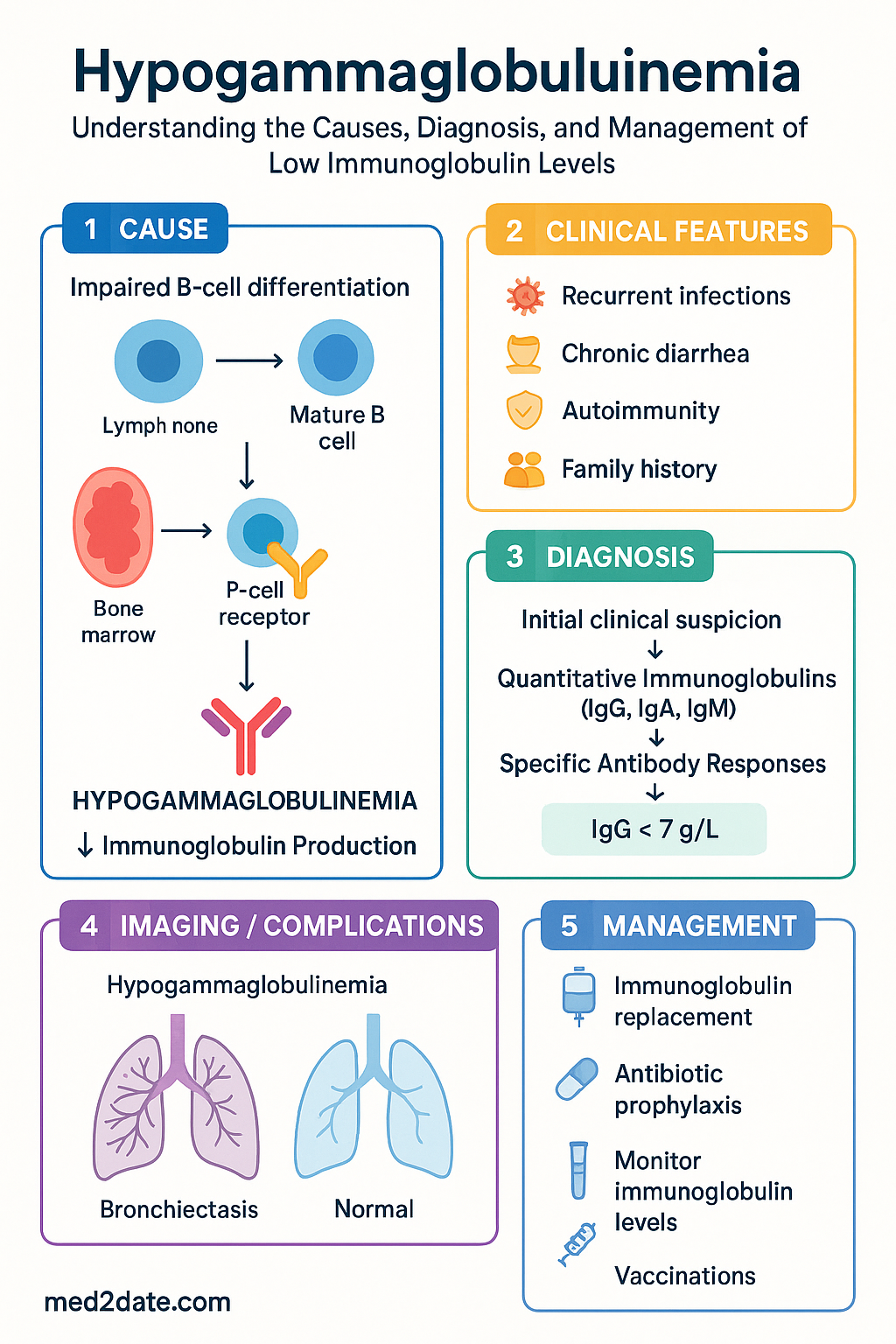
Causes of Hypogammaglobulinemia
Primary (Inherited) Causes
| Condition | Inheritance | Key Defect | Typical Onset | Immunoglobulin Pattern |
|---|---|---|---|---|
| Common Variable Immunodeficiency (CVID) | Usually sporadic; some autosomal dominant/recessive (ICOS, TACI, BAFF-R, CD19 mutations) | Impaired B-cell differentiation; reduced class-switched memory B cells | 2nd–4th decade (peak 20–40 years) | ↓ IgG ± ↓ IgA and/or ↓ IgB |
| X-linked Agammaglobulinemia (XLA / Bruton) | X-linked recessive (BTK gene, Xq22) | Block in pre-B to mature B-cell transition; absent mature B cells | 6–9 months (post-maternal IgG waning) | ↓↓↓ IgG, IgA, IgM (all isotypes) |
| X-linked Hyper-IgM Syndrome | X-linked (CD40LG / CD40L) | Defective T-cell CD40L–B-cell CD40 interaction; failed class switching | 1–2 years | ↓ IgG, ↓ IgA, ↑↑ IgM |
| Selective IgA Deficiency | Polygenic / multifactorial | Failed IgA class switching | Often asymptomatic; diagnosed at any age | ↓↓ IgA (<0.07 g/L); normal IgG, IgM |
| Transient Hypogammaglobulinemia of Infancy (THI) | Not inherited; developmental delay in IgG production | Delayed maturation of IgG synthesis | 6 months – 3 years (self-resolving by 3–5 years) | ↓ IgG (IgA, IgM often normal) |
| Hyper-IgM Syndrome (AID, UNG defects) | Autosomal recessive | Defective somatic hypermutation and class-switch recombination | Early childhood | ↓ IgG, ↓ IgA, ↑ IgM |
| Good Syndrome | Not inherited; associated with thymoma | B-cell lymphopenia and hypogammaglobulinemia with thymic tumour | 4th–6th decade | ↓ IgG ± ↓ IgA, IgM; low B cells |
Secondary (Acquired) Causes
Secondary hypogammaglobulinemia is considerably more common than primary immunodeficiency in general practice. The following table summarises major acquired aetiologies:
| Category | Examples | Mechanism |
|---|---|---|
| Haematological malignancy | CLL, multiple myeloma, non-Hodgkin lymphoma | Malignant clone displaces normal B cells; excess Ig catabolism |
| Protein loss | Nephrotic syndrome, protein-losing enteropathy (e.g. Ménétrier disease), severe burns | Urinary or GI loss exceeds hepatic synthesis rate |
| Immunosuppressive therapy | Rituximab (anti-CD20), high-dose corticosteroids, mycophenolate, cyclophosphamide | B-cell depletion or suppression of Ig synthesis |
| Anti-epileptic medications | Carbamazepine, phenytoin, valproate | Idiopathic suppression of IgA and/or IgG |
| Chronic infections | HIV, CMV, EBV, congenital rubella | Direct B-cell infection or immune dysregulation |
| Prematurity | Premature neonates <32 weeks | Inadequate transplacental IgG transfer |
| Malnutrition | Severe protein-calorie malnutrition | Substrate deficiency for Ig synthesis |
Clinical Features
The clinical presentation of hypogammaglobulinemia depends on the severity of Ig deficiency, the specific isotype(s) affected, and the underlying aetiology. Patients with primary antibody deficiency typically present in childhood (XLA, THI) or early adulthood (CVID), while secondary causes may present at any age.
Infectious Manifestations
| Site | Common Infections | Typical Organisms |
|---|---|---|
| Respiratory (upper) | Recurrent otitis media, sinusitis, mastoiditis | Streptococcus pneumoniae, Haemophilus influenzae (non-typeable) |
| Respiratory (lower) | Community-acquired pneumonia (≥1/year), bronchiectasis | S. pneumoniae, H. influenzae, Moraxella catarrhalis, Pseudomonas aeruginosa (late-stage bronchiectasis) |
| Gastrointestinal | Chronic diarrhoea, malabsorption, Giardia lamblia infection | Giardia lamblia, Campylobacter, norovirus, rotavirus (prolonged) |
| Skin / Soft tissue | Cellulitis, abscesses, infected eczema | Staphylococcus aureus, streptococci, Gram-negatives |
| CNS | Bacterial meningitis (encapsulated organisms) | S. pneumoniae, H. influenzae type b, Neisseria meningitidis |
| Septicaemia | Bacteraemia, sepsis | Encapsulated organisms; Gram-negatives (XLA) |
Non-Infectious Manifestations (particularly in CVID)
- Autoimmune disease: Immune thrombocytopenia (ITP), autoimmune haemolytic anaemia (AIHA), rheumatoid arthritis, pernicious anaemia, vitiligo — occur in 20–30% of CVID patients
- Granulomatous-lymphocytic infiltrative disease (GLILD): Non-caseating granulomas in lungs, liver, spleen, and lymph nodes mimicking sarcoidosis
- Splenomegaly: Present in 25–30% of CVID; may be massive
- Enteropathy: Coeliac-like villous atrophy, inflammatory bowel disease-like features, nodular lymphoid hyperplasia
- Bronchiectasis: Develops in 30–50% of CVID patients if IgRT is delayed; irreversible structural lung damage
- Malignancy: Increased risk of lymphoma (especially non-Hodgkin), gastric cancer (associated with Helicobacter pylori and achlorhydria)
Diagnosis
Stepwise Diagnostic Approach
Investigations Summary
Risk Stratification & Severity Scoring
Severity of hypogammaglobulinemia correlates with the degree of IgG reduction, functional antibody impairment, and clinical infection burden. Risk stratification guides urgency of IgRT initiation and monitoring intensity.
Empirical Antibiotic Therapy
While immunoglobulin replacement therapy (IgRT) is the definitive treatment, antibiotics play a critical adjunctive role. All patients should receive prompt empirical antibiotics for acute infections, and many require long-term prophylaxis.
Acute Infection Management
| Clinical Scenario | First-Line Agent | Second-Line / Allergy Alternative | Duration |
|---|---|---|---|
| Acute bacterial sinusitis / otitis media | Amoxicillin 500 mg PO TDS (adults) or 45 mg/kg/day PO BD (paediatric) | Amoxicillin-clavulanate 875/125 mg PO BD (adults); if penicillin allergy: azithromycin 500 mg day 1 then 250 mg days 2–5 (adults) or 10 mg/kg day 1 then 5 mg/kg days 2–5 (paediatric) | 10–14 days (longer course than immunocompetent) |
| Community-acquired pneumonia | Amoxicillin 1 g PO TDS (mild–moderate) OR benzylpenicillin 1.2 g IV 4–6 hourly (severe) | Ceftriaxone 1–2 g IV daily; add azithromycin 500 mg PO/IV daily if atypical co-infection suspected; if severe penicillin allergy: moxifloxacin 400 mg PO/IV daily | 10–14 days (oral step-down when clinically improving) |
| Bronchiectasis exacerbation | Amoxicillin 1 g PO TDS (if not known Pseudomonas) OR ciprofloxacin 500–750 mg PO BD (Pseudomonas suspected/confirmed) | Co-trimoxazole 960 mg PO BD; or meropenem 1 g IV TDS (hospitalised severe exacerbation) | 14 days |
| Giardiasis | Tinidazole 2 g PO single dose (adults) or 50 mg/kg (max 2 g) single dose (paediatric) | Metronidazole 400 mg PO TDS for 10 days (adults); nitazoxanide for refractory cases | Single dose (tinidazole) or 10 days (metronidazole) |
Antibiotic Prophylaxis
Long-term prophylactic antibiotics are recommended for patients with recurrent infections despite adequate IgRT, established bronchiectasis, or chronic sinopulmonary disease. Regimens should be guided by local AMR patterns and sputum culture results.
Immunoglobulin Replacement Therapy (IgRT)
Immunoglobulin replacement therapy is the definitive treatment for clinically significant hypogammaglobulinemia. In Australia, IgRT is governed by the National Blood Authority (NBA) Immunoglobulin Governance Programme, and prescription requires PBS Authority approval.
IVIG Products Available in Australia
Subcutaneous Immunoglobulin (SCIG)
SCIG is increasingly preferred in Australia due to the convenience of home self-administration, steady-state IgG levels, reduced systemic adverse effects, and improved quality of life — particularly for patients in regional and remote areas far from infusion centres.
IgRT Dosing Principles
- Target trough IgG ≥7 g/L: The primary therapeutic goal. Higher troughs (≥8–10 g/L) may be needed for patients with bronchiectasis, GLILD, or chronic lung disease.
- Starting dose: 400–600 mg/kg every 3–4 weeks (IV) or 100–200 mg/kg weekly (SC). Adjust based on trough levels and clinical response.
- Trough monitoring: Measure serum IgG immediately before the next scheduled dose. Check every 3–6 months initially, then at least annually once stable.
- IV to SC conversion: Multiply previous monthly IVIG dose by 1.37 and divide into weekly SC doses. Equivalent steady-state IgG levels are typically achieved.
- Lifelong therapy: IgRT is lifelong for primary antibody deficiency. Discontinuation is associated with recurrent infections and bronchiectasis progression.
Adverse Effects of IgRT
| Adverse Effect | Frequency | Management |
|---|---|---|
| Headache, myalgia, fever, chills | Common (5–15% of infusions) | Reduce infusion rate; pre-medicate with paracetamol, antihistamine ± hydrocortisone |
| Anaphylaxis / anaphylactoid | Rare (<1:10,000) | Stop infusion immediately; IM adrenaline 0.01 mg/kg (max 0.5 mg adults, 0.3 mg child); switch product or route to SC |
| Aseptic meningitis | Uncommon (1–3%) | Slow infusion rate; IV fluids; paracetamol/ibuprofen; may resolve with SCIG conversion |
| Thromboembolic events (DVT, PE, stroke) | Rare (risk factors: age >65, immobility, hyperviscosity, high infusion rate) | Slow infusion rate; hydrate; consider low-molecular-weight heparin prophylaxis in high-risk patients |
| Haemolysis (passive anti-A/anti-B) | Rare; more common with high-dose IVIG | Check FBC and haptoglobin post-infusion; monitor for delayed haemolysis (days 1–10) |
| Local site reactions (SCIG) | Common initially (15–30%); diminishes with time | Rotate sites; warm product to room temperature; use hyaluronidase if persistent |
Monitoring
Lifelong monitoring is essential to ensure adequate IgG trough levels, detect complications early (bronchiectasis, autoimmunity, lymphoproliferative disease), and optimise quality of life.
- Trough serum IgG (target ≥7 g/L)
- Infection diary review (frequency, severity, organisms)
- FBC, CRP/ESR, liver function, renal function
- Adherence assessment (especially SCIG self-administration)
- Trough serum IgG
- Spirometry (FEV₁, FVC) and DLCO
- Sputum culture (including mycobacterial culture if new infiltrates)
- Hepatitis B surface antibody (re-immunise if waning)
- Review antibiotic prophylaxis regimen
- Assessment for autoimmune complications (FBC, Coombs test, thyroid function)
- Nutritional status and growth (paediatric patients)
- High-resolution CT chest (HRCT) — for bronchiectasis surveillance
- Lymphocyte subsets if new cytopenias or lymphoproliferative features
- Gastroscopy with duodenal biopsy (if GI symptoms or malabsorption)
- Ultrasound abdomen (splenomegaly, lymphadenopathy)
- Lung function testing during acute exacerbations
- CT neck/chest/abdomen/pelvis if suspected lymphoma or GLILD
- Bone densitometry (DEXA) if chronic corticosteroid use
- Ophthalmology review if retinal complications suspected
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Australasian Society of Clinical Immunology and Allergy (ASCIA). Primary Immunodeficiency (PID) Guide for Health Professionals. ASCIA; 2024. Available at: https://www.ascia.org.au
- 2. National Blood Authority (NBA). National Policy on the Supply and Use of Human Immunoglobulin. Australian Government; 2023. Available at: https://www.blood.gov.au/ig
- 3. Bonilla FA, Khan DA, Ballas ZK, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol. 2015;136(5):1186–1205.e78.
- 4. Seidel MG, Kindle G, Gathmann B, et al. The European Society for Immunodeficiencies (ESID) Registry working definitions for the clinical diagnosis of inborn errors of immunity. J Allergy Clin Immunol Pract. 2019;7(6):1763–1770.
- 5. Chapel H, Lucas M, Lee M, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112(2):277–286.
- 6. Bruton OC. Agammaglobulinemia. Pediatrics. 1952;9(6):722–728.
- 7. Bousfiha AA, Jeddane L, Picard C, et al. The 2022 update of IUIS phenotypical classification for human inborn errors of immunity. J Clin Immunol. 2022;42(5):1008–1020.
- 8. Pharmaceutical Benefits Scheme (PBS). Immunoglobulin — Authority Required. Australian Government Department of Health; 2024. Available at: https://www.pbs.gov.au
- 9. Jolles S, Bernatowska E, de Gracia J, et al. Efficacy and safety of Hizentra® in patients with primary immunodeficiency after a dose-equivalent switch from intravenous or subcutaneous replacement therapy. Clin Immunol. 2011;141(1):90–102.
- 10. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander Health Performance Framework 2023 Summary Report. Canberra: AIHW; 2023.
- 11. Chang C, Roberts S, Shetty AK, et al. Management of bronchiectasis in Aboriginal and Torres Strait Islander peoples: an evidence review. Lung Foundation Australia. 2022.
- 12. Lucas M, Lee M, Lortan J, Lopez-Granados E, Misbah S, Chapel H. Infection outcomes in patients with common variable immunodeficiency disorders: relationship to immunoglobulin therapy over 22 years. J Allergy Clin Immunol. 2010;125(6):1354–1360.e4.
- 13. Peters L, Bousfiha A, de Vries E, et al. International Consensus Document (ICON): common variable immunodeficiency. J Clin Immunol. 2016;36(Suppl 1):34–41.
- 14. National Health and Medical Research Council (NHMRC). The Australian Immunisation Handbook. 11th ed. Australian Government Department of Health; 2022 (updated 2024). Available at: https://immunisationhandbook.health.gov.au
- 15. Kainulainen L, Nikoskelainen J, Vuorinen T, et al. Viruses and bacteria in bronchial samples from patients with primary hypogammaglobulinemia. Am J Respir Crit Care Med. 1999;159(4):1199–1204.