📋 Key Information Summary
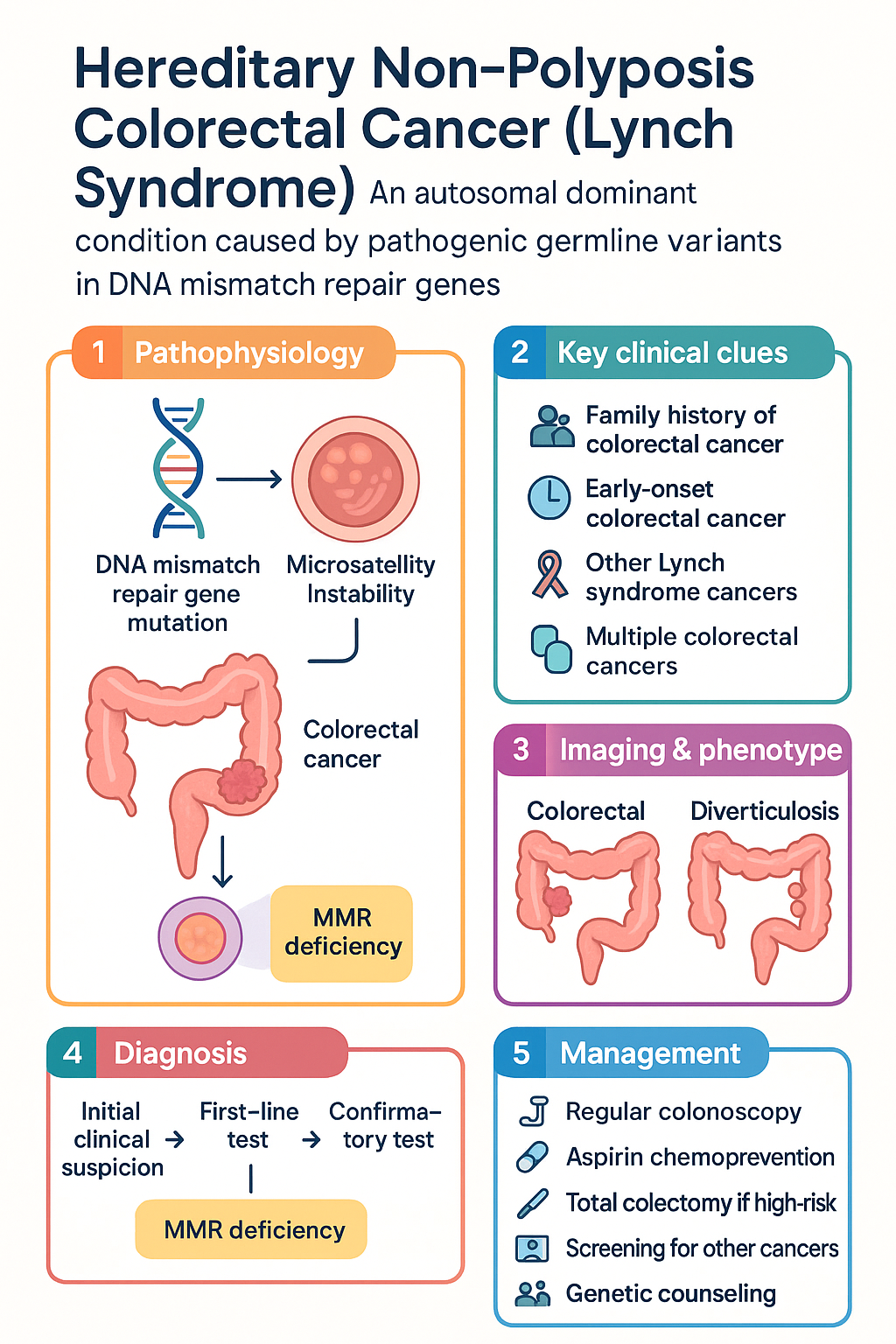
- Lynch syndrome (hereditary non-polyposis colorectal cancer) is an autosomal dominant condition caused by pathogenic germline variants in DNA mismatch repair (MMR) genes — MLH1, MSH2, MSH6, or PMS2 — or EPCAM deletions silencing MSH2.
- Accounts for approximately 3–5 % of all colorectal cancers and 2–3 % of endometrial cancers in Australia.
- Lifetime colorectal cancer risk is 15–52 % depending on the specific gene affected; MLH1/MSH2 carriers face the highest risk.
- Extracolonic cancers include endometrial (up to 60 %), ovarian, gastric, urinary tract, hepatobiliary, small bowel, brain (turcot variant), and sebaceous neoplasms (Muir–Torre variant).
- Universal tumour testing — immunohistochemistry (IHC) for MMR protein expression and/or microsatellite instability (MSI) — is recommended for all colorectal and endometrial cancers at diagnosis to identify Lynch syndrome.
- The Amsterdam II criteria (clinical) and Bethesda guidelines (tumour-testing trigger) help identify families warranting genetic referral and testing.
- Surveillance colonoscopy should commence at age 25 years or 2–5 years before the earliest CRC in the family, whichever is earlier, repeated every 1–2 years.
- Surveillance for extracolonic cancers — including upper GI endoscopy, urinalysis, gynaecological screening — is recommended from age 30–35 years.
- Aspirin chemoprevention (≥ 300 mg/day for ≥ 2 years) reduces CRC incidence by approximately 35–40 % based on the CAPP2 trial; current NHMRC-endorsed guidance supports consideration of aspirin 100–300 mg daily.
- Prophylactic hysterectomy with bilateral salpingo-oophorectomy (BSO) should be discussed with female carriers from age 35–40 years on completion of childbearing.
- Lynch-associated colorectal cancers are frequently microsatellite instability–high (MSI-H) and may respond to immune checkpoint inhibitors (pembrolizumab) if metastatic and refractory.
- Genetic testing and counselling are available through family cancer clinics across all Australian states and territories, with Telehealth access for remote communities.
Introduction & Australian Epidemiology
Lynch syndrome (LS), historically termed hereditary non-polyposis colorectal cancer (HNPCC), is the most common inherited colorectal cancer predisposition syndrome. It follows an autosomal dominant pattern of inheritance with high penetrance, caused by pathogenic germline variants in the DNA mismatch repair (MMR) genes: MLH1, MSH2, MSH6, and PMS2, as well as deletions in the EPCAM gene that epigenetically silence MSH2.
The MMR system corrects single-nucleotide mismatches and insertion–deletion loops that arise during DNA replication. When MMR function is lost, cells accumulate replication errors — particularly at microsatellite repeat sequences — a phenomenon termed microsatellite instability (MSI). This genomic instability drives tumourigenesis in colorectal epithelium and other tissues.
In Australia, Lynch syndrome is estimated to affect approximately 1 in 280–370 individuals, implying there are roughly 70,000–90,000 carriers nationally. Despite this prevalence, fewer than 5 % of carriers have been identified — the remainder remain undiagnosed and unscreened, representing a significant public health gap.
The Cancer Council Australia and the Australasian Society of Gastrointestinal Pathologists (ASGIP) recommend universal tumour testing — MMR immunohistochemistry (IHC) and/or MSI testing — for every newly diagnosed colorectal and endometrial cancer. This strategy has been shown to be cost-effective in the Australian healthcare setting and is now standard practice in most major cancer centres.
| Parameter | Estimate |
|---|---|
| Population prevalence | 1 in 280–370 individuals |
| Proportion of all CRC attributable to LS | 3–5 % |
| Proportion of all endometrial cancer | 2–3 % |
| Diagnosed carriers in Australia | < 5 % of total estimated carriers |
| Median age at CRC diagnosis (untreated) | 44–61 years (gene-dependent) |
Genetics — MLH1, MSH2, MSH6, PMS2 & EPCAM
Lynch syndrome is caused by monoallelic (heterozygous) pathogenic germline variants in one of four core MMR genes. A somatic "second hit" — typically loss of heterozygosity, promoter methylation, or a second somatic mutation — inactivates the remaining allele and triggers the MMR-deficient phenotype. The specific gene involved determines penetrance, age of onset, and the spectrum of associated cancers.
| Gene | Chromosome | Protein | % of LS families | Lifetime CRC risk | Lifetime endometrial risk | Key features |
|---|---|---|---|---|---|---|
| MLH1 | 3p22.2 | MutL homolog 1 | ~40 % | 41–52 % | 14–33 % | Highest penetrance; earliest CRC onset; most common in Australian LS families |
| MSH2 | 2p21 | MutS homolog 2 | ~34 % | 33–52 % | 21–60 % | High endometrial risk; Muir–Torre syndrome (sebaceous tumours); associated with EPCAM deletions |
| MSH6 | 2p16.3 | MutS homolog 6 | ~18 % | 15–33 % | 16–44 % | Later-onset CRC; particularly high endometrial cancer risk; may be missed by MSI testing alone |
| PMS2 | 7p22.1 | PMS1 homolog 2 | ~8 % | 15–20 % | 12–15 % | Lowest penetrance; later onset; pseudogene complicate molecular testing; compound heterozygosity causes constitutional MMR deficiency (CMMRD) in childhood |
| EPCAM deletions | 2p21 | Epithelial cell adhesion molecule | ~1–3 % | Up to 75 % by age 70 | Up to 12 % | 3′ deletions cause promoter hypermethylation of adjacent MSH2 through transcriptional read-through |
Immunohistochemistry Patterns
Loss of nuclear staining on IHC for the respective MMR protein(s) guides targeted germline testing. The characteristic IHC patterns are:
| IHC loss pattern | Likely germline gene | Notes |
|---|---|---|
| Loss of MLH1 + PMS2 | MLH1 | Must exclude somatic BRAF V600E mutation or MLH1 promoter hypermethylation (sporadic MSI-H CRC) |
| Loss of MSH2 + MSH6 | MSH2 or EPCAM | Highly specific for LS; proceed directly to MSH2/EPCAM germline testing |
| Isolated MSH6 loss | MSH6 | MSI may be low or stable; rely on IHC rather than MSI panel alone |
| Isolated PMS2 loss | PMS2 | Beware pseudogene interference; specialised long-range PCR or NGS recommended |
Constitutional MMR Deficiency (CMMRD)
Biallelic pathogenic variants in MMR genes (most commonly PMS2) cause CMMRD, a severe childhood cancer predisposition syndrome characterised by brain tumours, haematological malignancies, and early-onset CRC. Suspect CMMRD in children with café-au-lait macules, childhood malignancy, and family history suggestive of Lynch syndrome. Diagnosis requires referral to a specialised familial cancer centre.
Amsterdam II & Bethesda Criteria
Two sets of clinical criteria help identify individuals and families warranting further investigation for Lynch syndrome. The Amsterdam II criteria are clinical/familial, while the revised Bethesda guidelines indicate which tumours should undergo MMR testing.
Amsterdam II Criteria (1999)
All of the following must be met:
- Three or more relatives with an LS-associated cancer (colorectal, endometrial, small bowel, ureter, or renal pelvis)
- One affected relative is a first-degree relative of the other two
- Two or more successive generations affected
- One or more cancers diagnosed before age 50 years
- Familial adenomatous polyposis excluded
- Tumours verified by histopathology
Revised Bethesda Guidelines (2004)
Tumour testing (MSI or MMR IHC) is recommended for colorectal cancers when any one of the following applies:
- CRC diagnosed in a patient aged < 50 years
- Synchronous or metachronous LS-associated tumour (colorectal, extracolonic) regardless of age
- CRC with MSI-H histology (tumour-infiltrating lymphocytes, Crohn-like reaction, mucinous/signet-ring differentiation, medullary pattern) diagnosed before age 60 years
- CRC diagnosed in one or more first-degree relatives with an LS-associated tumour, with one cancer diagnosed before age 50 years
- CRC diagnosed in two or more first- or second-degree relatives with LS-associated tumours at any age
Australian Current Practice
The Cancer Council Australia, the Clinical Oncology Society of Australia (COSA), and the Royal College of Pathologists of Australasia (RCPA) now endorse universal tumour testing for all CRC and endometrial cancers regardless of age or family history. This supersedes the age and family-history triggers in the Bethesda guidelines and is supported by cost-effectiveness analyses in the Australian setting.
Cancer Risks & Surveillance
Lynch syndrome confers elevated lifetime risks of multiple cancer types. The risk magnitude varies by gene. The following table summarises cumulative lifetime risks from pooled cohort studies.
| Cancer type | MLH1 | MSH2 | MSH6 | PMS2 |
|---|---|---|---|---|
| Colorectal | 41–52 % | 33–52 % | 15–33 % | 15–20 % |
| Endometrial | 14–33 % | 21–60 % | 16–44 % | 12–15 % |
| Ovarian | 4–20 % | 8–38 % | ~10 % | ~3 % |
| Gastric | ~5 % | ~5 % | ~5 % | ~5 % |
| Urinary tract | ~4 % | 7–12 % | ~2 % | ~2 % |
| Small bowel | ~4 % | ~6 % | ~1 % | ~1 % |
| Brain/CNS | ~1 % | ~7 % | ~1 % | ~1 % |
Surveillance Protocol
The following surveillance schedule is based on the Cancer Council Australia Clinical Practice Guidelines for Surveillance Colonoscopy, the European Hereditary Tumour Group (EHTG) 2024 guidelines, and the Australian Family Cancer Clinics consensus.
| Organ / Test | Commence age | Interval | Notes |
|---|---|---|---|
| Colonoscopy | 25 years or 2–5 years before earliest family CRC | Every 1–2 years | If MSH6/PMS2, may consider starting at age 30; annual if prior adenoma or CRC |
| Upper GI endoscopy + duodenoscopy | 30–35 years | Every 3–5 years | More frequent if family history of gastric cancer; consider H. pylori eradication |
| Urinalysis + urine cytology | 30–35 years | Annually | Especially for MSH2 carriers; urine cytology + imaging for haematuria |
| Gynaecological review ± TVU ± CA-125 | 30–35 years | Annually | Discuss prophylactic surgery; screening has limited evidence for mortality reduction |
| Consider abdominal ultrasound | 30–35 years | Every 1–2 years | For hepatobiliary and small bowel surveillance in high-risk genotypes |
| Skin examination | At diagnosis | Annually | Especially for MSH2 — Muir–Torre syndrome (sebaceous gland tumours) |
Management & Prophylactic Surgery
Management of Lynch syndrome encompasses cancer prevention (chemoprevention and prophylactic surgery), intensive surveillance, treatment of established cancers, and cascade testing of at-risk relatives.
Aspirin Chemoprevention
The CAPP2 randomised controlled trial demonstrated that aspirin 600 mg daily for at least 2 years reduced CRC incidence by approximately 35–40 % in LS carriers. The CAPP3 trial is ongoing, investigating lower doses (100 mg, 300 mg, 600 mg).
Prophylactic Surgery
Prophylactic Gynaecological Surgery
Prophylactic hysterectomy with bilateral salpingo-oophorectomy (BSO) should be discussed with all female LS carriers from age 35–40 years, or on completion of childbearing. This approach eliminates endometrial cancer risk and substantially reduces ovarian cancer risk. There is no reliable screening method for early detection of ovarian cancer in LS.
Prophylactic Colectomy
Prophylactic total colectomy is not routinely recommended for LS carriers without CRC, given the effectiveness of colonoscopic surveillance and adenoma removal. However, prophylactic (procto-)colectomy may be considered in specific circumstances:
- Inability to undergo adequate colonoscopic surveillance
- Multiple unresectable flat adenomas or failed endoscopic management
- MLH1 or MSH2 carriers with high anxiety about CRC risk after shared decision-making
Treatment of LS-Associated Colorectal Cancer
LS-associated CRCs have distinct biological features that influence treatment:
- Stage II CRC: Excellent prognosis (~95 % 5-year survival); 5-fluorouracil (5-FU) adjuvant chemotherapy is NOT recommended as it provides no additional benefit and may cause harm in MMR-deficient stage II tumours.
- Stage III CRC: Adjuvant FOLFOX (folinic acid + 5-FU + oxaliplatin) is standard; the survival benefit is similar to sporadic CRC.
- Metastatic CRC: MSI-H tumours are highly responsive to immune checkpoint inhibitors. Pembrolizumab (Keynote-177) and nivolumab ± ipilimumab (CheckMate 142) are first-line options for MSI-H/dMMR metastatic CRC.
Cascade Genetic Testing
Once a pathogenic MMR variant is identified in a proband, all first-degree relatives should be offered targeted predictive genetic testing. This is the single most impactful intervention in LS management — identifying carriers before cancer develops enables surveillance and prevention.
In Australia, cascade testing is funded through public familial cancer clinics. Telehealth and postal saliva collection kits are available for geographically remote families.
Special Populations
Pregnancy
Paediatrics
Elderly (> 75 years)
Renal Impairment
Hepatic Impairment
Immunocompromised
Aboriginal & Torres Strait Islander Health Considerations
📚 References
- 1. Vasen HFA, Mecklin J-P, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum. 1991;34(5):424–425.
- 2. Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96(4):261–268.
- 3. Burn J, Gerdes A-M, Macrae F, et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet. 2011;378(9809):2081–2087.
- 4. Engel C, Ahadova A, Seppälä TT, et al. Associations of pathogenic variants in MLH1, MSH2, and MSH6 with risk of colorectal adenomas and tumors and with somatic mutations in patients with Lynch syndrome. Gastroenterology. 2020;158(5):1326–1333.
- 5. Cancer Council Australia. Clinical Practice Guidelines for Surveillance Colonoscopy. Sydney: Cancer Council Australia; 2018. Available from: wiki.cancer.org.au.
- 6. Australian Institute of Health and Welfare (AIHW). Cancer in Aboriginal & Torres Strait Islander people of Australia. Cat. no. CAN 109. Canberra: AIHW; 2018.
- 7. Royal College of Pathologists of Australasia (RCPA). MMR Immunohistochemistry and Microsatellite Instability Testing for Lynch Syndrome: Quality Assurance Programme. Sydney: RCPA; 2022.
- 8. Guidelines NG12: Colorectal cancer. National Institute for Health and Care Excellence (NICE). Updated 2020. Available from: nice.org.uk/guidance/ng12.
- 9. Monahan KJ, Bradshaw N, Dolwani S, et al. Guidelines for the management of hereditary colorectal cancer from the British Society of Gastroenterology (BSG)/Association of Coloproctology of Great Britain and Ireland (ACPGBI)/United Kingdom Cancer Genetics Group (UKCGG). Gut. 2020;69(3):411–444.
- 10. Stoffel EM, Mangu PB, Gruber SB, et al. Hereditary colorectal cancer syndromes: American Society of Clinical Oncology Clinical Practice Guideline endorsement of the familial risk–colorectal cancer: European Society for Medical Oncology Clinical Practice Guidelines. J Clin Oncol. 2015;33(2):209–217.
- 11. Biller LH, Syngal S, Yurgelun MB. Recent advances in Lynch syndrome. Fam Cancer. 2019;18(2):211–219.
- 12. André T, Shiu K-K, Kim TW, et al. Pembrolizumab versus chemotherapy for microsatellite instability–high or mismatch repair–deficient metastatic colorectal cancer (KEYNOTE-177): final analysis of a randomised, open-label, phase 3 study. Lancet Oncol. 2022;23(5):659–670.
- 13. Crosbie EJ, Ryan NAJ, Arends MJ, et al. The Manchester International Consensus Group recommendations for the management of gynaecological cancers in Lynch syndrome. Genet Med. 2019;21(10):2390–2400.