📋 Key Information Summary
- Familial adenomatous polyposis (FAP) is an autosomal dominant condition caused by germline pathogenic variants in the APC tumour-suppressor gene on chromosome 5q21–22; approximately 70–80 % of cases are inherited and 20–30 % arise from de novo mutations. class="guideline-li">Classic FAP is characterised by the development of hundreds to thousands of colorectal adenomatous polyps, typically by the mid-teens, with virtually 100 % lifetime risk of colorectal carcinoma (CRC) if left untreated — median age of CRC diagnosis ~39 years.
- Attenuated FAP (AFAP), associated with mutations at the extreme 5′ or 3′ ends of APC, presents with fewer polyps (<100), later onset, and CRC risk around 70 % by age 80.
- The genotype–phenotype correlation guides clinical management: codon 1250–1464 mutations predict profuse polyposis and earlier need for surgery; codon 1–157 and >1595 mutations correlate with AFAP.
- Extracolonic manifestations are common and include upper-gastrointestinal polyps (fundic gland, adenomatous), desmoid tumours, hepatoblastoma, thyroid carcinoma, CNS tumours (Turcot variant), and congenital hypertrophy of the retinal pigment epithelium (CHRPE).
- Screening for APC mutations should be offered to all first-degree relatives of known FAP patients; predictive genetic testing is recommended from age 10–12 years in classic FAP families.
- Colectomy remains the cornerstone of CRC prevention in classic FAP — options include total proctocolectomy with ileal pouch–anal anastomosis (IPAA), or total abdominal colectomy with ileorectal anastomosis (IRA) in selected patients with low rectal polyp burden.
- Chemoprevention with sulindac or celecoxib can reduce polyp number and size but has NOT demonstrated prevention of CRC and is adjunctive only; celecoxib 400 mg BD is PBS Authority Required for FAP.
- Lifelong endoscopic surveillance of the upper and lower gastrointestinal tract is mandatory — annual flexible sigmoidoscopy after IRA, ileal-pouch surveillance after IPAA, and upper-GI endoscopy every 1–3 years from age 25.
- Desmoid tumours are the leading cause of morbidity and second leading cause of mortality in FAP; management ranges from observation (asymptomatic) to sulindac plus tamoxifen, or cytotoxic chemotherapy for aggressive disease.
- Aboriginal and Torres Strait Islander Australians have lower rates of genetic testing and referral for hereditary CRC syndromes; culturally safe outreach and community-based genetic counselling can improve uptake of surveillance and prophylactic surgery.
- A multidisciplinary team involving gastroenterology, colorectal surgery, clinical genetics, oncology, endocrinology (thyroid screening), ophthalmology, and psychology is essential for optimal FAP care.
Introduction & Australian Epidemiology
Familial adenomatous polyposis (FAP) is the most common hereditary colorectal cancer syndrome, accounting for approximately 1 % of all colorectal carcinomas worldwide. It is an autosomal dominant condition caused by inactivating germline pathogenic variants in the APC (adenomatous polyposis coli) tumour-suppressor gene located on chromosome 5q21–22. In the classic form, hundreds to thousands of adenomatous polyps carpet the colorectal mucosa beginning in adolescence; without prophylactic colectomy, progression to CRC is virtually inevitable, with a median age at diagnosis of approximately 39 years.
In Australia, FAP affects an estimated 1 in 7,000–10,000 live births, with approximately 250–400 affected individuals identified at any given time. The Australian Institute of Health and Welfare (AIHW) reports that hereditary non-polyposis colorectal cancer (HNPCC/Lynch syndrome) and FAP together account for 5–10 % of CRC cases nationally. However, under-diagnosis remains a significant issue, particularly in regional, rural, and remote communities, and among Aboriginal and Torres Strait Islander peoples, where access to genetic testing and specialist gastroenterology services is limited.
The Cancer Genetics Clinic network — including services at the Peter MacCallum Cancer Centre (Melbourne), Familial Cancer Service (Sydney), Genetic Health Queensland, and the South Australian Clinical Genetics Service — coordinates most Australian FAP registries. National guidelines from Cancer Australia and the eviQ resource provide evidence-based management pathways. This guideline synthesises current evidence on genetics, clinical features, extracolonic manifestations, and surveillance/management strategies applicable to the Australian healthcare setting.
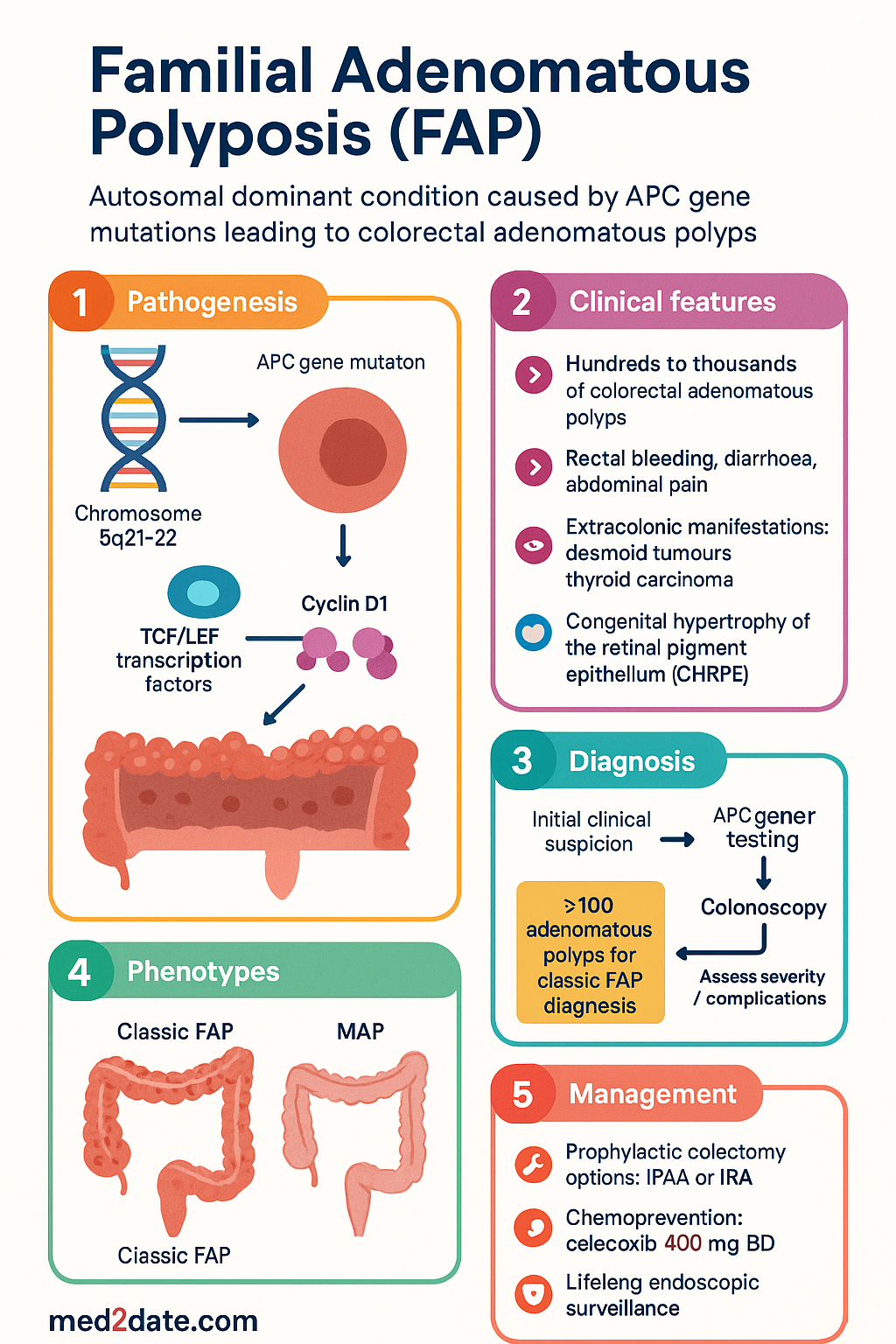
Genetics & Pathogenesis
The APC Gene and the Wnt Signalling Pathway
The APC gene encodes a 2,843-amino-acid protein that functions as a critical negative regulator of the canonical Wnt/β-catenin signalling pathway. In normal colonic epithelium, the APC protein forms a destruction complex with Axin, glycogen synthase kinase-3β (GSK-3β), and casein kinase 1α, promoting phosphorylation and proteasomal degradation of β-catenin. When APC is inactivated by germline mutation, β-catenin accumulates in the nucleus, constitutively activating TCF/LEF transcription factors and driving uncontrolled expression of pro-proliferative genes such as MYC and Cyclin D1.
Germline Mutations
Approximately 70–80 % of FAP patients carry a germline pathogenic variant in APC. The mutational spectrum includes:
- Nonsense and frameshift variants (~70 %): Premature termination codons leading to truncated, non-functional protein; the majority cluster in exon 15 (codons 1,250–1,464).
- Splice-site variants (~10 %): Aberrant mRNA splicing with variable phenotypic severity.
- Large deletions / rearrangements (~5–10 %): Detected by MLPA (multiplex ligation-dependent probe amplification) when sequencing is negative; may account for a higher proportion of apparent de novo cases.
In approximately 20–30 % of patients, the pathogenic variant is de novo (no family history), underscoring the importance of genetic testing even in apparently sporadic polyposis.
Genotype–Phenotype Correlations
| Mutation Region | Codon Range | Phenotype |
|---|---|---|
| 5′ end (exon 4–9) | Codon 1–157 | Attenuated FAP; fewer polyps, later onset |
| Central cluster (exon 15) | Codon 158–495 | Classic FAP; CHRPE; desmoid risk may be lower |
| Mutation cluster region | Codon 1,250–1,330 | Profuse polyposis (>5,000 polyps); earliest CRC risk |
| Codon 1,309 | Codon 1,309 | Most severe; CRC ~10 years earlier than average |
| 3′ end | Codon >1,595 | Attenuated FAP; higher desmoid risk |
| Desmoid-associated | Codon 1,395–2,000 | 50–80 % desmoid risk; "Turcot" overlap |
The Multi-Hit (Knudson) Model
FAP exemplifies the Knudson two-hit hypothesis: the inherited first hit (germline APC mutation) is followed by somatic loss of the remaining wild-type allele (loss of heterozygosity, LOH) in individual colonic epithelial cells. Additional somatic mutations in KRAS, SMAD4, and TP53 then drive the adenoma→carcinoma sequence at an accelerated rate. The APC mutation is termed the "gatekeeper" of colorectal tumorigenesis.
MYH-Associated Polyposis (MAP) — Key Differential
MUTYH-associated polyposis (MAP) is an autosomal recessive condition caused by biallelic pathogenic variants in MUTYH, a base-excision repair gene. It phenotypically mimics attenuated FAP but is distinguished by autosomal recessive inheritance, absence of APC mutations, and characteristic somatic APC G:C→T:A transversions. All patients with multiple adenomas and negative APC testing should undergo MUTYH analysis.
Classic & Attenuated FAP Features
Classic FAP
Classic FAP is defined by the presence of ≥100 colorectal adenomatous polyps detectable by flexible sigmoidoscopy or colonoscopy, typically manifesting by age 15–20 years. If untreated, virtually 100 % of patients develop CRC, with a median age of CRC onset of 39 years (range 34–43). Key clinical features include:
- Hundreds to thousands of 2–10 mm adenomatous polyps carpeting the colorectum, with density greatest in the rectum and sigmoid.
- Symptomatic presentation may include rectal bleeding, diarrhoea, mucus discharge, abdominal pain, or iron-deficiency anaemia.
- Polyps begin as early as age 10; CRC has been reported as early as age 15 in high-risk genotypes (codon 1309).
- Without intervention, almost all patients will require colectomy by their mid-20s.
Attenuated FAP (AFAP)
Attenuated FAP is characterised by a milder phenotype with fewer colorectal polyps (typically 10–99), later onset (mean age at diagnosis ~35–45 years), and a CRC risk of approximately 70 % by age 80 if not managed. AFAP is associated with mutations at the extreme 5′ end of APC (exons 4–9, codons 1–157) or at the 3′ end (codons >1,595). Key distinctions from classic FAP:
| Feature | Classic FAP | Attenuated FAP |
|---|---|---|
| Polyp number | ≥100 (often >1,000) | 10–99 (sometimes <10) |
| Age at onset | 10–20 years | 20–45 years |
| CRC risk | ~100 % | ~70 % by age 80 |
| Polyp distribution | Diffuse; rectal predominance | Right-sided predominance; sparse in rectum |
| Mutation site | Codon 158–1,464 | Codon 1–157 or >1,595 |
| Surgical approach | Prophylactic colectomy in late teens | Surveillance-guided; colectomy may be deferred |
| CHRPE | Common | Rare or absent |
| Desmoid risk | Moderate (10–15 %) | Lower (unless 3′ mutation) |
Gardner Syndrome and Turcot Syndrome
Gardner syndrome and Turcot syndrome are phenotypic variants of FAP — not separate genetic entities:
- Gardner syndrome: FAP plus extra-intestinal benign tumours — osteomas (mandible, skull), epidermoid cysts, desmoid tumours, dental abnormalities (unerupted teeth, supernumerary teeth), and CHRPE.
- Turcot syndrome: FAP plus central nervous system tumours — most commonly medulloblastoma (APC-related) or glioblastoma (more often associated with mismatch repair gene mutations / Lynch syndrome). APC-related Turcot carries a median age of brain tumour diagnosis of ~14 years.
Extracolonic Manifestations
FAP is a true multi-organ syndrome. Awareness of extracolonic manifestations is essential for comprehensive surveillance and early intervention. The following table summarises the major manifestations, estimated prevalence, and recommended screening approach.
| Organ System | Manifestation | Prevalence in FAP | Screening / Management |
|---|---|---|---|
| Upper GI | Duodenal / periampullary adenomas | 50–90 % | Upper GI EGD from age 25; Spigelman staging every 1–3 years |
| Upper GI | Fundic gland polyps | 30–88 % | Biopsy if >10 mm or dysplastic |
| Gastric | Gastric adenomas | 5–10 % (higher in Asian FAP) | Endoscopic resection; surveillance |
| Soft tissue | Desmoid tumours | 10–25 % (up to 80 % with 3′ mutations) | MRI surveillance; surgical excision for symptomatic / enlarging lesions |
| Thyroid | Papillary thyroid carcinoma | 2–12 % (female predominance) | Annual thyroid ultrasound from age 15 |
| Hepatobiliary | Hepatoblastoma | 1.6 % (mostly children <5 years) | Serum AFP every 6 months from birth to age 15 |
| CNS | Medulloblastoma (Turcot) | <1 % | Low threshold for neuroimaging if neurological symptoms |
| Ophthalmic | Congenital hypertrophy of the retinal pigment epithelium (CHRPE) | 60–80 % (classic FAP) | Baseline fundoscopy; no treatment needed (diagnostic marker) |
| Dental | Osteomas, supernumerary teeth, unerupted teeth | 20–75 % | Panoramic dental X-ray; oral surgery review |
| Adrenal | Adrenal adenomas / carcinomas | 7–13 % | Incidental on CT; functional work-up if >4 cm |
Duodenal Adenomas and Spigelman Classification
Duodenal adenocarcinoma is the second most common malignancy in FAP after CRC, carrying a cumulative lifetime risk of 4–12 %. The Spigelman staging system (Stage 0–IV) guides surveillance intervals and intervention thresholds:
Desmoid Tumours
Desmoid tumours (aggressive fibromatosis) are the leading cause of morbidity and the second leading cause of death in FAP patients. They arise in approximately 10–25 % of FAP patients overall (up to 80 % in those with 3′ APC mutations) and are most common in the mesentery, abdominal wall, and extra-abdominal sites. Risk factors for desmoid development include prior abdominal surgery, female sex, and positive family history of desmoids.
Surveillance & Management
Genetic Testing and Cascade Screening
Genetic testing for the known familial APC pathogenic variant should be offered to all first-degree relatives of confirmed FAP patients. Predictive genetic testing in children is recommended from age 10–12 years (classic FAP) or 18–20 years (AFAP), preceded by age-appropriate genetic counselling. Mutation-negative relatives can be discharged from intensive surveillance.
Colorectal Surveillance
Surgical Options
| Procedure | Description | Advantages | Disadvantages | Indication |
|---|---|---|---|---|
| TAC + IRA | Total abdominal colectomy with ileorectal anastomosis | Better bowel function; lower surgical morbidity; no permanent stoma | Rectal remnant retains CRC risk (0.7–1 %/year); requires lifelong surveillance | Classic FAP with <20 rectal polyps and no rectal cancer |
| TPC + IPAA | Total proctocolectomy with ileal pouch–anal anastomosis (J-pouch) | Removes virtually all at-risk mucosa; eliminates rectal cancer risk | Higher morbidity; pouchitis (up to 50 %); reduced fertility in women; worse stool frequency | Classic FAP with >20 rectal polyps; rectal cancer; AFAP with severe rectal involvement |
| TPC + IRA / end ileostomy | Total proctocolectomy with end ileostomy | Lowest residual cancer risk | Permanent stoma; significant QoL impact | When IPAA not feasible (low rectal cancer, sphincter incompetence, severe desmoid) |
Upper Gastrointestinal Surveillance
All FAP patients should commence upper GI endoscopy (EGD) from age 25 (or 20 in high-risk genotypes). The Spigelman classification guides ongoing intervals:
- Spigelman Stage 0–I: EGD every 4–5 years
- Spigelman Stage II: EGD every 2–3 years
- Spigelman Stage III: EGD every 1–2 years; consider endoscopic mucosal resection (EMR) or argon plasma coagulation
- Spigelman Stage IV: EGD every 6–12 months; surgical consultation for pancreas-sparing duodenectomy or Whipple procedure
Thyroid Surveillance
Thyroid ultrasound is recommended annually from age 15 (some guidelines suggest from age 10). Papillary thyroid carcinoma in FAP is more common in women, tends to be multifocal, and may present at a younger age than sporadic thyroid cancer. Baseline thyroid function tests and neck ultrasound should be performed at the time of FAP diagnosis.
Hepatoblastoma Screening
For children with a known APC mutation or at 50 % risk (untested children of FAP patients): serum alpha-fetoprotein (AFP) and abdominal ultrasound every 6 months from birth until age 15. Hepatoblastoma is rare (~1.6 %) but treatable if detected early.
Chemoprevention
NSAIDs and COX-2 inhibitors can reduce the number and size of colorectal adenomas in FAP but have NOT been shown to prevent CRC or replace surgery:
Emerging Therapies
Several novel approaches are under investigation for FAP, including:
- Eflornithine ± sulindac: Ornithine decarboxylase inhibitor combined with sulindac has shown significant reduction in duodenal polyp progression in recent Phase III trials (Sulindac and Eflornithine in Treating Patients with Familial Adenomatous Polyposis — NCT01483144).
- Wnt pathway inhibitors: Porcupine inhibitors (e.g., ETC-159, WNT974) target Wnt ligand secretion; early-phase trials ongoing.
- Encapsulated faecal microbiota transplantation: Preliminary data suggest microbiome modulation may influence polyp growth.
None of these therapies are currently PBS-listed or approved by the TGA for FAP. Patients should be referred to clinical trials where appropriate.
Special Populations
Aboriginal and Torres Strait Islander Health Considerations
📚 References
- 1. Vasen HFA, Möslein G, Alonso A, et al. Guidelines for the clinical management of familial adenomatous polyposis (FAP). Gut. 2008;57(5):704–713. doi:10.1136/gut.2007.136127
- 2. Syngal S, Brand RE, Church JM, et al. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015;110(2):223–262. doi:10.1038/ajg.2014.435
- 3. >Monahan KJ, Bradshaw N, Dolwani S, et al. Guidelines for the management of hereditary colorectal cancer from the British Society of Gastroenterology (BSG)/Association of Coloproctology of Great Britain and Ireland (ACPGBI)/United Kingdom Cancer Genetics Group (UKCGG). Gut. 2020;69(3):411–444. doi:10.1136/gutjnl-2019-319915
- 4. Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis. 2009;4:22. doi:10.1186/1750-1172-4-22
- 5. Australian Institute of Health and Welfare (AIHW). Cancer in Australia 2021. Cancer series no. 133. Cat. no. CAN 144. Canberra: AIHW; 2021.
- 6. Cancer Australia. Clinical guidance for the management of colorectal cancer. Surry Hills, NSW: Cancer Australia; 2021.
- 7. Church J, Simmang C. Practice parameters for the treatment of patients with dominantly inherited colorectal cancer (familial adenomatous polyposis and hereditary nonpolyposis colorectal cancer). Dis Colon Rectum. 2003;46(8):1001–1012.
- 8. Spigelman AD, Williams CB, Talbot IC, et al. Upper gastrointestinal cancer in patients with familial adenomatous polyposis. Lancet. 1989;2(8666):783–785. doi:10.1016/S0140-6736(89)90840-8
- 9. Nieuwenhuis MH, Lefevre JH, Bülow S, et al. Family history, surgery, and APC mutation are risk factors for desmoid tumors in familial adenomatous polyposis: an international cohort study. Dis Colon Rectum. 2011;54(10):1229–1234.
- 10. >Giardiello FM, Hamilton SR, Krush AJ, et al. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med. 1993;328(18):1313–1316. doi:10.1056/NEJM199305063281805
- 11. Steinbach G, Lynch PM, Phillips RKS, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. 2000;342(26):1946–1952. doi:10.1056/NEJM200006293422603
- 12. Australian Institute of Health and Welfare (AIHW). Aboriginal and Torres Strait Islander health performance framework 2020 summary report. Canberra: AIHW; 2020.
- 13. Cancer Council Australia. Clinical practice guidelines for the prevention, early detection and management of colorectal cancer. Sydney: Cancer Council Australia; 2017.
- 14. Burke W, Petersen G, Lynch P, et al. Recommendations for follow-up care of individuals with an inherited predisposition to cancer. I. Hereditary nonpolyposis colon cancer. JAMA. 1997;277(11):915–919.
- 15. Koskenvuo L, Pitkäniemi J, Rantanen M, et al. Impact of screening on survival in familial adenomatous polyposis. J Clin Gastroenterol. 2016;50(1):35–40.